
A multicellular system isn’t a system just because it has many cells. The cells must be of different types. Indeed, a great many different cell types are needed. To distinguish as many cell types as possible, researchers have resorted to various techniques, including morphological analysis, fluorescence-activated cell sorting, and RNA sequencing. However, the ultimate technique for distinguishing cells of different types may be single-cell mass spectrometry (MS) proteomics.
No other method is as comprehensive. It can, for example, capture the post-translational modifications that add the final layer of complexity to gene expression. Moreover, it collects information with single-cell resolution, uncovering biological complexities beyond the reach of bulk tissue analysis. Ultimately, it can reveal, like no other technique, the degree to which multicellular “systemicity” depends on cellular heterogeneity.
Converging on diversity
To catch up on the latest in single-cell MS proteomics, GEN spoke with Nikolai Slavov, PhD, associate professor of bioengineering at Northeastern University, director of the Single-Cell Proteomics Center at Northeastern, and an Allen Distinguished Investigator. He noted that several advances are “coming to fruition together [and] making it a wonderful time to be doing MS proteomics.”

Northeastern University
“Advances in sample preparation are improving sensitivity, throughput, and depth of proteome coverage,” Slavov detailed. These improvements, he added, support activities such as the “analysis of individual mammalian cells, parallel analysis of proteins by data-independent acquisition, and combined simultaneous analysis of proteins.” Overall, advances in MS proteomics are giving, in Slavov’s estimation, “multiplicative increases in throughput and corresponding decreases in cost.”
Slavov’s laboratory has developed new methods for high-throughput single-cell MS proteomics. They include Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) and a more fully automated version of SCoPE-MS called SCoPE2. In recent works, Slavov and colleagues reported that SCOPE-MS was used to quantify proteome heterogeneity during cell differentiation1 and that SCoPE2 was used to quantify the emergence of macrophage heterogeneity.2
It should be emphasized that advances in single-cell MS proteomics are not limited to identifying proteins or measuring their abundances. Innovations are being extended to measure protein functions (such as enzymatic activities), native conformations, and protein-protein interactions. For example, covalent protein painting, an MS-based protein footprinting method used to survey protein folding throughout the proteome,3 has uncovered misfolded proteins in the Alzheimer’s proteome beyond the established culprits (that is, beyond misfolded b-amyloid and hyperphosphorylated tau).
Slavov credited covalent protein painting to the Scripps Research laboratory led by John R. Yates III, PhD, a professor of molecular medicine. “[This method] allows covalent labeling of exposed lysines on the surface of proteins,” Slavov remarked. “Upon subsequent MS, it allows protein conformations to be inferred at very high throughput and scale. And it’s not limited to isolated proteins as in traditional structure-based analyses—it can be done in living cells.” In a recent perspective article, Slavov described how the technique can be applied to the structural analyses of proteins in single cells.4
Addressing intercellular diversity
Single-cell proteomics methods, like all single-cell methods, respect the structural and functional diversity of cells. These methods preserve the biological meaning embedded in healthy and pathological samples instead of losing it by effectively averaging readings across cell populations. These methods transcend the limitations of bulk methods.
“[With bulk methods, we] take samples, mash up the different cell types, and measure the average protein levels,” Slavov remarked. “We end up with results that do not meaningfully represent human biology.” Clearly, bulk methods are especially limiting when one is studying multicellular organisms and hoping to understand how interactions among cells of different types can ensure the integrity of the whole.
Slavov is particularly excited about the prospect of analyzing variations across proteomes of single cells generated through “natural experiments” such as evolution and reproduction.5 “Single-cell analysis has enabled the inference of direct protein interactions and causal mechanisms between proteins,” he explained. “That allows us to [conduct more detailed studies of] how information is transmitted within signaling cascades in diverse biological systems.” He notes that when single-cell analysis was unavailable, it was necessary to “make assumptions based on convenience rather than reason.”
Label-free and multiplexed single-cell proteomics
For decades, the mainstay of specific, single-cell protein analysis has been the use of affinity regents such as aptamers, fused fluorophores, or antibodies. To detect proteins, investigators have used antibodies individually or in multiplexed panels, or fused them to rare earth metals (in studies using cytometry and time of flight, or CyTOF, technology) or DNA oligonucleotides (in studies using cellular indexing of transcriptomes and epitopes by sequencing, or CITE-seq, technology). However, such methods require specific antibodies that must be rigorously validated.
Single-cell MS proteomics offers the advantage of analyzing many more proteins in individual cells without the time, expense, or validations needed for antibodies. It avoids the risk that normal protein function will be disrupted through the use of an unwieldy fluorescent tag, such as green fluorescent protein. And it adopts label-free or multiplexed approaches, both of which benefit from improvements in sample preparation and peptide separation.
“The main advantage of multiplexed approaches is the ability to analyze many cells simultaneously, which increases throughput and reduces the cost of analysis per cell,” Slavov emphasized. “The advantages of label-free approaches include the ability to conduct studies with very few cells and to avoid artifacts associated with labeling. However, many of the exciting problems for single-cell proteomics require the analysis of very large numbers of cells.”
Slavov’s laboratory is focusing on developing single-cell workflows that offer high throughput and support the analysis of large numbers of cells. The laboratory maintains that these workflows should be economical and easily accessible to the entire MS proteomics research community.
Multiplexing relaxes time constraints
Protocols for single-cell MS proteomics can detect peptides with very high sensitivity. However, these protocols also need to deal with time constraints. Multiplexing and innovations in data acquisition and interpretation can relax these time constraints and increase proteome coverage.6
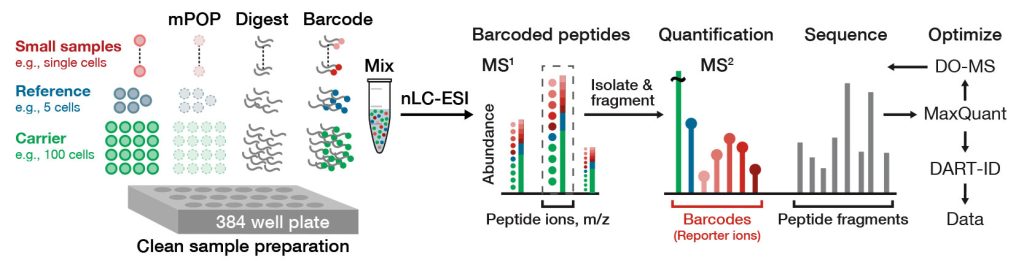
Multiplexed single-cell proteomics workflows have predominantly relied on isobaric mass tags, in combination with the isobaric carrier approach introduced by SCoPE-MS and maintained in other methods including SCoPE2. Chemical tags with identical masses (isobaric mass tags), where the distribution of the heavy isotopes varies in the tag’s structure, have enabled deep quantitative analyses of proteomes in small samples.
“There’s great potential in multiplexing with non-isobaric mass tags as well,” Slavov said. “In some ways, these tags are simpler, and they can provide additional advantages by enabling simultaneous multiplexing of peptide and cell analysis, which can multiplicatively increase throughput.”
Non-isobaric mass tags have different numbers of isotopes in the molecule. Peptides labeled in different samples with different mass tags ionize and separate identically in chromatography or electrophoresis. “This is wonderful because we can control for artifacts due to separation, ionization, etc.,” Slavov pointed out.
In MS, non-isobaric mass tags can give peptides different numbers of neutrons, allowing the peptides to be readily distinguished. Indeed, the use of non-isobaric mass tags enables multiplexing; however, using these tags with data-dependent acquisition poses a disadvantage. Peptides need to be analyzed one by one, prolonging analysis times and constraining the number of peptides that can be analyzed.
With data-dependent acquisition, peptide features are analyzed one peptide at a time. A peptide feature is a pattern of mass-to-charge peaks formed by different ions with different isotopes generated from a peptide. A single MS run may include hundreds of thousands of peptide features, and their number increases proportionately with the number of non-isobaric labels. Consequently, if the same depth of proteome coverage is to be achieved, more time must be spent analyzing peptide features when the number of labels increases. “Non-isobaric multiplexing fell out of favor because it imposed time constraints that resulted in lower proteome coverage,” Slavov remarked.
This led to the innovation and widespread adoption of isobaric multiplexing, where one can add the same number of neutrons to each mass tag but attach them to different places in the molecule. Initially, peptides labeled with different isobaric tags have the same mass, which results in a single peak, reducing the need to analyze an unwieldy number of peptide features. Upon isolation and fragmentation of peptide ions, isobaric mass tags fragment in a way that enables their identification and relative quantitation. The drawback of using isobaric mass tags, however, is the generation of impure spectra that lower accuracy of peptide detection.
“Increasing the time needed to analyze peptides using non-isobaric mass tags creates a problem only if one is analyzing one peptide feature at a time,” Slavov reiterated. “This cannot achieve deep proteome coverage and high throughput in a finite time because we need hundreds of milliseconds to collect enough ions per peptide. We cannot do that for 100,000 peptides to get deep coverage of the proteome of a cell.
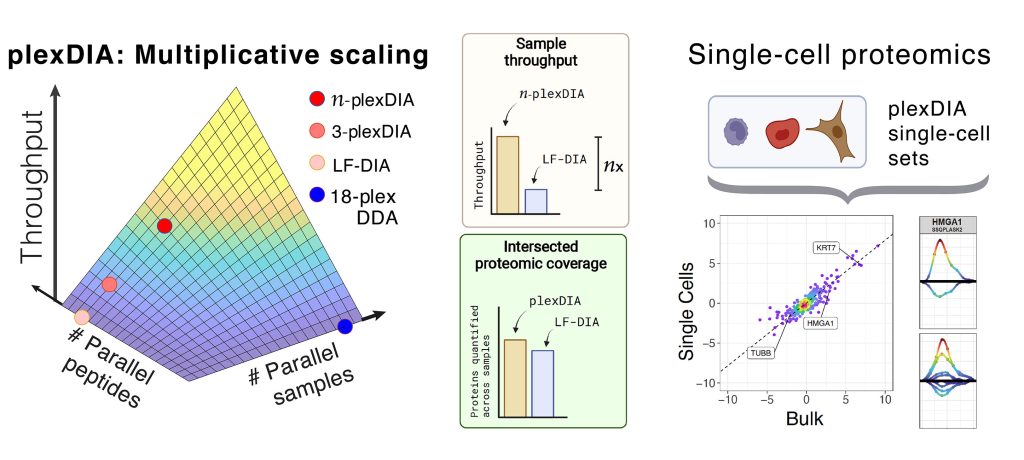
“Data-independent acquisition (DIA) methods allow broad isolation of many peptide features at a time, and these features can be analyzed in parallel. [These methods allow us] to multiplex both proteins and samples at the same time.”
A method recently developed by Slavov’s team uses non-isobaric mass tags to achieve deeper and more accurate proteome coverage. This method, which is called plexDIA, also avoids the problems one encounters in using isobaric mass tags and increasing throughput.7
The ability to analyze entire proteomes in many individual cells simultaneously through multiplexing and parallel analysis creates synergies due to natural structures in the acquired data. These can be exploited by computational algorithms to enhance peptide sequence identification.
Challenges in sample preparation
Sample preparation in single-cell MS proteomics presents a range of difficulties. Collection techniques need to be optimized so they can reproducibly include one cell per sample. Contaminants need to be eliminated. And processes need to be scaled up.
Developing methods to isolate one cell at a time with high quality would seem a simple task, Slavov observed. After all, it would seem that one could rely on flow cytometry, which is widely available and commonly used to isolate single cells. Also, MS instruments are exquisitely sensitive and capable of detecting unlabeled ions of all kinds. Unfortunately, these ions include contaminants. MS analysis of minute samples, such as single cells, need proactive methods to keep contaminants to a minimum.
Another difficulty arises when one processes conventional bulk samples. Because these samples often use chemicals that are incompatible with MS analysis, one must prepare clean samples that use only MS-compatible reagents, or one must remove incompatible reagents from samples. “While cleanup measures can be applied to bulk samples, the losses associated with cleanup can affect single-cell analysis much more substantially and adversely,” Slavov cautioned. “Developing procedures that use only MS-compatible agents has been a priority for my laboratory.”
When methods of preparing individual samples have been optimized, the challenge is to scale them up so they can accommodate thousands of individual single-cell samples in parallel. Methods amenable to scale up have been developed by other researchers. For example, researchers at Pacific Northwest National Laboratory showed that single-cell samples could be held in the miniwells of microfabricated chips.8
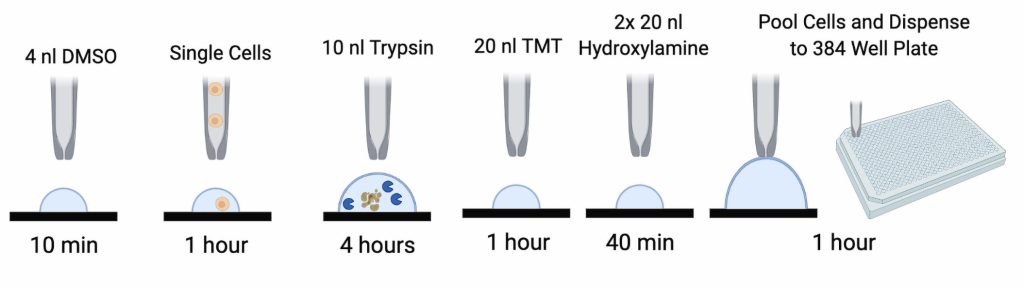
“We’ve developed a method called nano-ProteOmic sample Preparation, or nPOP,” Slavov added. “It uses an instrument to deposit tiny 20-nL droplets with high precision on a fluorocarbon-coated glass slide.” This method allows Slavov’s team to prepare over 2,000 single-cell samples simultaneously, and it can potentially be scaled up further.9
Genomics, transcriptomics, and single-cell proteomics can be synergistic; that is, when they are integrated with each other, their combined strength can exceed the sum of their separate strengths. Synergistic multiomics techniques are already reaping fresh biological insights, partly through innovations in sample preparation that allow proteins, RNA, and metabolites to be isolated at the same time. One approach analyzes peptides in one section of chromatography, and hydrophobic metabolites in another. Slavov anticipates that it will soon be possible “to analyze metabolites, proteins, and RNAs from the same single cell.”
Accessibility of single-cell methods
Some single-cell methods and instruments are widely available; others, less so. For example, methods that accomplish single-cell isolation via fluorescence-activated cell sorting are widely available at most research facilities. These methods can process a few hundred single-cell evaluations at a time. Alternatively, there are cutting-edge methods that can process thousands. The cutting-edge methods, however, typically require MS instrumentation. Unfortunately, even commercial MS instrumentation can be expensive.
For example, the nPOP technology developed by Slavov’s laboratory requires a commercial instrument that is too expensive to be broadly available. “That raises the bar for those who can use it,” Slavov admitted. “The least accessible protocols are those that require proprietary technology developed in the laboratory of an investigator.”
He added that even if the most cutting-edge technologies are out of reach, there are single-cell MS protocols that just about any laboratory can implement immediately. His own laboratory recently published a detailed protocol that uses “relatively inexpensive, broadly available equipment” for a SCoPE2 workflow.10
Although Slavov emphasized that although single-cell proteomics is becoming more technologically advanced, it still requires well-planned experimental designs. Even the most advanced technologies can generate useless data if experimental designs fail to account for confounding batch and biological effects.
References
1. Budnik B, Levy E, Harmange G, Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018; 19(1): 161. DOI: 10.1186/s13059-018-1547-5.
2. Specht H, Emmott E, Petelski AA, et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021; 22(1): 50. DOI: 10.1186/s13059-021-02267-5.
3. Bamberger C, Pankow S, Martínez-Bartolomé S, et al. Protein Footprinting via Covalent Protein Painting Reveals Structural Changes of the Proteome in Alzheimer’s Disease. J. Proteome Res. 2021; 20(5): 2762–2771. DOI: 10.1021/acs.jproteome.0c00912.
4. Slavov N. Measuring Protein Shapes in Living Cells. J. Proteome Res. 2021; 20(6): 3017. DOI: 10.1021/acs.jproteome.1c00376.
5. Slavov N. Learning from natural variation across the proteomes of single cells. PLoS Biol. 2022; 20(1): e3001512. DOI: 10.1371/journal.pbio.3001512.
6. Slavov N. Driving Single Cell Proteomics Forward with Innovation. J. Proteome Res. 2021; 20(11): 4915–4 918. DOI: 10.1021/acs.jproteome.1c00639.
7. Derks J, Leduc A, Wallmann G, et al. Increasing the throughput of sensitive proteomics by plexDIA. Nat Biotechnol. 2022. DOI: 10.1038/s41587-022-01389-w.
8. Huang C-M, Zhu Y, Jin DQ, et al. Direct Surface and Droplet Microsampling for Electrospray Ionization Mass Spectrometry Analysis with an Integrated Dual-Probe Microfluidic Chip. Anal. Chem. 2017; 89(17): 9009–9016. DOI: 10.1021/acs.analchem.7b01679.
9. Leduc A, Huffman RG, Cantlon J, et al. Exploring functional protein covariation across single cells using nPOP. bioRxiv 2022. DOI: 10.1101/2021.04.24.441211.
10. Petelski AA, Emmott E, Leduc A, et al. Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 2021; 16(12): 5398–5425. DOI: 10.1038/s41596-021-00616-z.