Categorization has preoccupied biologists going back to the days of classical Greece, when no less a figure than Aristotle classified living things by asking successive narrowing questions such as, “Is it animal or vegetable?” and “How many legs does it have?”
At times, biological categorization has gone awry, resulting in famous gaffes (Aristotle himself said that spiders had six legs and thus qualified as insects)—yet the practice has endured.
More recently, biological categorization was taken up by Charles Darwin, who distinguished between splitters and lumpers. Splitters, he said, are “those who make many species,” whereas lumpers are “those who make few.”
Today, categorization is reaching ever deeper into the stuff of life. For example, it is establishing categories based on differences in gene expression that occur from cell to cell. To find these differences, latter-day Aristotles and Darwins are relying on a new technique: RNA sequencing, or RNA-seq. It promises, as did earlier exercises in categorization, to reveal new entities and previously unknown relationships among them. For example, it may distinguish cell subtypes that are more or less significant at different stages of development, or in different states of health and disease.
Originally, RNA-seq was more of a lumper. It began with bulk RNA-seq approaches, which measure average gene expression levels across cell populations. Increasingly, however, RNA-seq is becoming more of a splitter. The ultimate in transcriptome-level splitting is single-cell RNA-seq technology.
Bulk RNA-seq and single-cell RNA-seq technologies are both capable of providing deep, rapid, and unbiased analyses of the transcriptome, and both are becoming routine in the study of gene expression. Single-cell RNA-seq, however, surpasses bulk RNA-seq in terms of the kinds of information it can generate. It is single-cell RNA-seq that can identify novel cell types, characterize tumor heterogeneity, and follow the cell-fate decisions that shape development. If, however, single-cell RNA-seq is to deliver all these advantages, it must be backed by specialized methods of data analysis.
Normalizing Single-Cell Results
“Like many other groups, up until about two years ago, our group was using a method from bulk analysis for RNA-seq normalization,” says Christina Kendziorski, Ph.D., professor of biostatistics and medical informatics, University of Wisconsin–Madison. “We eventually noticed that this method did not work well for normalizing single-cell RNA-seq data.”
One of the goals in data normalization is to remove technical artifacts, which include features such as sequencing depth, gene length, or GC content. In bulk RNA-seq, an increase in sequencing depth leads to an almost directly proportional increase in expression, and this relationship between sequencing depth and expression is common across all genes.
Consider a weakly expressed gene. Here, a doubling of the sequencing depth would lead to an average doubling in expression. Moreover, for a highly expressed gene, the same relationship would be maintained.
“For single-cell RNA-seq, we noticed that this relationship was not common across genes,” recalls Dr. Kendziorski. In the case of single-cell RNA-seq, highly expressed genes appeared similar to the genes captured by bulk sequencing methods in terms of the count-depth relationship, which expresses how the sequencing counts behave as sequencing depth is increased. “However, the moderately and weakly expressed genes,” Dr. Kendziorski continues, “did not track with sequencing depth as expected.”
Therefore, when methods from bulk normalization were used, highly expressed genes were normalized correctly, but moderately and weakly expressed genes were over-normalized. “Consequently, for these genes,” notes Dr. Kendziorski, “normalization methods from bulk RNA-seq applied in the single-cell setting introduced artifacts.”
In a recent study, Dr. Kendziorski and colleagues introduced SCnorm, a computational method that estimates the dependence of transcript expression on sequencing depth for every gene and allows single-cell RNA-seq data to be normalized accurately and efficiently. “We expect that SCnorm will lead to substantial improvements in downstream inference in a number of areas,” asserts Dr. Kendziorski.
What’s next for computational methods in single-cell RNA-seq? “There is a big opportunity to develop methods for network analysis in single-cell data,” remarks Dr. Kendziorski, who concedes that such analysis is still uncommon. “The methods from bulk RNA-seq, even if we tweak them a little bit, will lose some information—potentially a lot of information.”
There is also a critical need for methods that integrate data from multiple single-cell technologies, such as those that profile DNA, expression, methylation, and so on. “The technologies are here,” observes Dr. Kendziorski, “but we do not yet have robust computational methods to integrate single-cell data across multiple sources.”
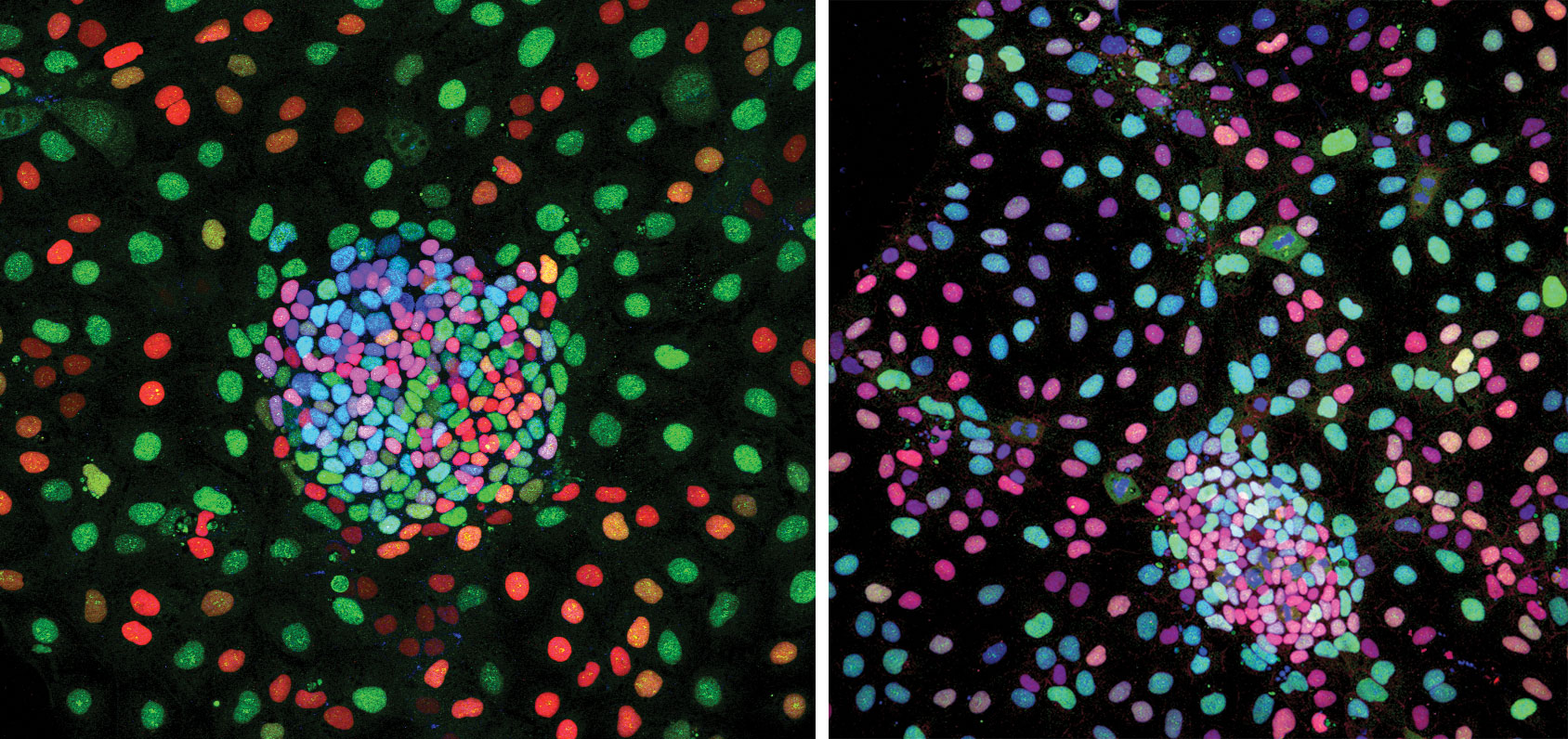
Capturing Morphological Context
Although single-cell RNA-seq offers more finely grained information than does bulk RNA-seq, the newer technology poses unique technical difficulties. For example, there is potential for error in the isolation of single cells. Even if single-cell RNA-seq succeeds in identifying distinct cell types, it may fail to preserve spatial information, that is, information about where cells of a given type occur within intact tissue.
To correlate transcripts of interest with morphological features, scientists at Advanced Cell Diagnostics developed the RNAscope® technology, an in situ hybridization assay that can detect target RNA with cell specificity in intact tissues. The RNAscope technology amplifies target-specific signals while simultaneously suppressing nonspecific hybridization that would lead to background noise.
“For any candidate genes of interest identified by RNA-seq, we can easily look in intact tissue in context of pathology, to see where that gene is expressed,” asserts Christopher Bunker, Ph.D., vice president of business development, Advanced Cell Diagnostics. “Using an in situ method for RNA detection enables verification of a transcript’s cell of origin.”
“In this assay,” he continues, “we use a pool of oligonucleotide probes, which we can design for specific detection of any transcript of interest, any splice variant, and even mutations identified as being interesting or relevant from RNA sequencing. These are used with a universal set of our RNAscope amplification and detection reagents.”
According to Dr. Bunker, the method is sensitive, specific, and resolves single RNA molecules within intact cells and tissues. It can be applied to any gene of interest, and it can be multiplexed with cell markers up to 4-plex. “The assay is done on intact tissue,” Dr. Bunker notes, “but it resembles real-time PCR in terms of its simplicity and sensitivity.”
One of the applications of the RNAscope assay is to validate findings from single-cell RNA-seq studies. “Wider awareness and adoption of RNAscope in situ hybridization (ISH) as a cross-validation method might reduce the need to perform single-cell analyses,” explains Dr. Bunker. “One could very quickly go from identifying any transcript of interest (for instance, a gene that is upregulated in disease) to determining which cells in intact tissue are expressing that gene.”
He adds that because RNAscope and RNA-seq are both capable of delivering results in a very time-efficient manner, they may be performed sequentially. Specifically, the techniques may be used together to examine the targets discovered through sequencing so as to prioritize them. That is, the targets most relevant to disease may be identified.
“We have another newly developed RNA ISH assay, a variation called BaseScope,” informs Dr. Bunker. “It can detect deletion and point mutations in intact tissue.” BaseScope is an ISH assay that is based on the same platform as the RNAscope technology. According to Dr. Bunker, BaseScope promises value not only for the verifying and validating of mutations discovered by RNA-seq, but also for interrogating the heterogeneity of those mutations within the tumor tissue.
“We have used BaseScope for detection of about a dozen mutations so far, such as KRAS, BRAF, and EGFR,” says Dr. Bunker. While RNA-seq can identify those mutations in tumor tissue, it does not necessarily indicate whether they occur homogeneously, throughout the tumor, or whether they represent heterogeneous phenomena. “By doing ISH,” insists Dr. Bunker, “it is possible to obtain an actual view of which cells have the mutation versus the wild-type copy.”
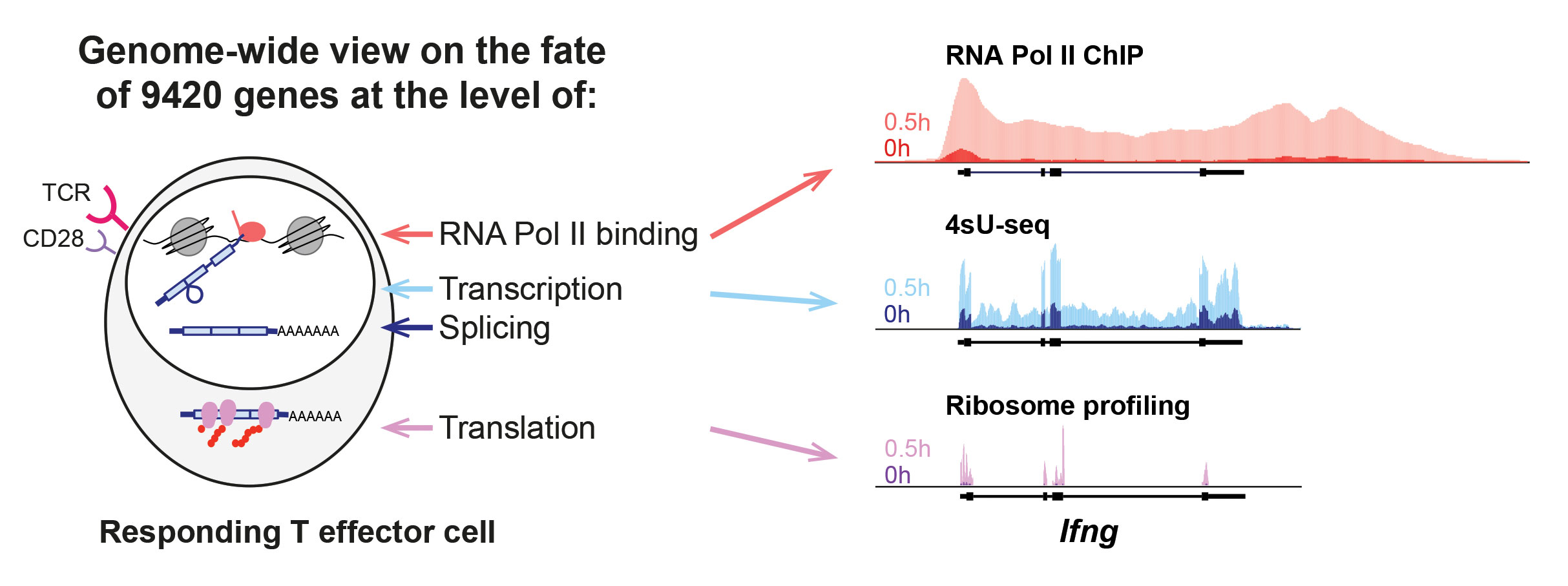
Tracking Temporal Dynamics
“We performed a real-time analysis during the primary T helper (Th)-cell response, tracking changes from RNA polymerase II binding to changes in the transcriptome and translatome,” says Elke Glasmacher, Ph.D., group leader, Institute of Diabetes and Obesity, Helmholtz Zentrum München. She adds that her team saw this analysis as a way of working toward a display of the “overall comprehensive course of events.”
It has been known for a long time that the activation of immune cells results in rapid functional changes. Details about the underlying dynamics of the gene expression events, however, have been more elusive.
In a study that combined time courses of 4-thiouridine sequencing, RNA-seq, ribosome profiling, and RNA polymerase II chromatin immunoprecipitation sequencing (ChIP-seq), Dr. Glasmacher and colleagues examined the genome-wide temporal dynamics of transcriptional and post-transcriptional changes in approximately 10,000 genes during T-cell activation. This study was the first comprehensive real-time analysis of the temporal dynamics of transcription, splicing, and translation during effector T-cell activation.
The analysis revealed that transcription and translation are highly coupled for ~92% of the genes, and it showed that for a few genes, transcription and translation are independently regulated. Gene upregulation mostly occurred by the rapid de novo recruitment of RNA Pol II to genetic loci, and the release of paused RNA Pol II from promoter-proximal positions played only a minor role, particularly for immediate-early genes.
“Our findings provide a foundation for immunologists to identify previously unnoticed genes that are highly regulated,” declares Dr. Glasmacher. These genes, she adds, “might in the end also play a role in certain disease contexts.”
The study also revealed very different intrinsic translation rates for some of the genes. “For example, it appears that cytokine mRNAs have a different translation rate than other genes,” Dr. Glasmacher points out. “We did not analyze this in depth yet, but it will be the foundation for future studies.”
When transcription and translation rates of activated T cells were compared with the rates found in nonactivated cells, the researchers discovered there were few differences. However, among activated T cells, intrinsic cell turnover and translation rates differed, resulting in differences in the level of gene expression downstream. Also, these rates did not change to a great extent for most genes.
“One thing that fascinates us are the amounts of noncoding transcripts that appear in cells,” says Dr. Glasmacher. “From a biological perspective, it is of interest to find out what they are doing. From a technical point of view, it is still challenging to map all the sequences with certainty.”
Revisiting Immune Cell Taxonomies
Cellular markers can be trusted only so far, suggests Nir Hacohen, Ph.D., associate professor of medicine at Harvard Medical School and member of the Broad Institute. They are, he explains, of uncertain utility as markers of pure cell types.
Over the years, many of the cellular markers that have been identified have been used to divide cell types into subtypes. “But every time we use one of these markers, we cannot know for sure whether the cellular subtype we found is pure or not,” complains Dr. Hacohen.
Even if markers are used that appear to be ideal for separating cellular populations into subpopulations, it is often challenging to determine whether a subpopulation consists of one cell type (and is “pure”), or whether it represents a mixture of multiple cell types. “We have been using these markers for years—not because we thought the practice was perfect, but because this was the way we could do it,” notes Dr. Hacohen.
The classification of cells has been particularly challenging when the cells to be subtyped are dendritic cells or monocytes. Historically, these cells were defined based on morphology, physiology, localization, development, and surface markers.
Rather than rely on marker-based assays to identify cellular subpopulations, Dr. Hacohen and colleagues tried a different approach. They used deep sequencing at the single-cell level and unbiased clustering. This approach, the scientists found, proved capable of revealing new types of human blood dendritic cells, monocytes, and progenitors.
The scientists performed single-cell RNA-seq for about 2,400 cells enriched for HLA-DR+ cells isolated from healthy blood donors. As a result of this work, Dr. Hacohen and colleagues defined six dendritic cell and four monocyte populations. In addition, they found that one of the dendritic cell subtypes, DC5, exists in a continuum, or a set of states that are similar to each other, but not identical.
“I look at this approach as a new microscope that allows us to see structures that we did not see before,” declares Dr. Hacohen. In that sense, single-cell RNA-seq differs markedly from bulk RNA-seq. “Bulk RNA-seq provides biomarkers and hypotheses for pathways,” he points out, “whereas single-cell RNA-seq basically takes away the ambiguity from bulk RNA-seq, where one assumes that everybody is doing the same thing because there is no other choice.”
Even though the single-cell RNA-seq approach is unbiased, and even though it is capable of identifying rare cell types as well as the relationships between cell types, it may still miss some cell types. “There could be very low-level RNAs that are not captured even with single-cell RNA-seq,” admits Dr. Hacohen, “and that would indicate that more complexity exists.” Additionally, this approach would miss cell types that are defined by metabolites or non-RNA molecules.
“In the future, it will be important to understand and settle differences between cellular states and their functional relevance,” predicts Dr. Hacohen. “While this will take a lot of work, we imagine that it will happen in the coming years.”
A better understanding of the cellular subpopulations will help us visualize the continuum of states that can exist. It may also, Dr. Hacohen suggests, help us link continuums of states with different disease states and pathogenicity. Tackling these “fundamental questions” was hard even to imagine, he notes, “before these methods existed.”
Parallel Single-Cell Sequencing
Human pluripotent stem cells (hPSCs), with their ability for infinite self-renewal, have the potential to be an unlimited source of cells for a variety of biomedical applications, ranging from drug screening to cell-replacement therapy and in vitro organogenesis. Many of these applications typically require reproducible generation of a large number of target cells. This is where research efforts hit a speed bump, as stem cell cultures are often heterogeneous.
Maroof Adil, Ph.D., a postdoctoral researcher in Professor David Schaffer’s lab at UC Berkeley, uses single-cell RNA-seq to better understand population heterogeneity within hPSCs cultured in vitro. His lab develops 3D biomaterials to increase the efficiency of the large-scale production of clinical-grade stem cells and the mature cells that are derived from them. These biomaterials could help expand and differentiate hPSCs into neurons, ultimately to decipher the complicated process of neurodevelopment to treat central nervous system diseases such as Parkinson’s and Huntington’s.
Single-cell RNA-seq is a powerful technology that provides a high-resolution snapshot of the population distribution within a biological sample. Using a single-cell sequencing technology jointly developed by Illumina and Bio-Rad, Dr. Adil’s team explores heterogeneity of hPSCs. The system isolates and barcodes thousands of single cells in a matter of minutes using a droplet-partitioning technology. The cells are then directed to downstream sequencing. The time from a single-cell suspension to single cells and barcoded beads in droplets is about five minutes, allowing researchers to handle sensitive primary cells that can die quickly during separation.
Conventionally, cells are isolated by flow cytometry, RNA extracted individually, and sometimes even sequenced individually as well. By contrast, the Berkeley team says it drastically reduces time spent in sample preparation and data collection. Dr. Adil hopes that using this technology to better understand the population heterogeneity and dynamics within pluripotent stem cell cultures may allow enhanced control over stem cell fate, and facilitate biomedical applications of stem cell technology.
Single-Cell RNA-Seq
RNA-seq has become one of the standard tools for gene expression, displacing legacy methods such as hybridization arrays. Single-cell RNA-seq has recently evolved as a powerful method to resolve sample heterogeneity and reveal hidden biology that may be missed in bulk RNA-seq. However, single-cell experiments require considerable expertise for effective experimental design, execution, and data analysis.
Initial single-cell RNA-seq experiments were costly, often 10-fold more expensive than traditional bulk methods. A number of strategies have been employed to create cost-effective, high-throughput methods to drastically bring down the cost of single-cell experiments.
“Fluorescence-activated cell sorting (FACS) has emerged as a leading platform for cell-sorting and capture because it is fast and enables high-throughput processing of a heterogeneous mixture of cells to enrich the most important cells,” says Eleen Shum, Ph.D., a scientist at BD Genomics. “The workflow of RNA sequencing using BD FACS to isolate cells starts from a single-cell suspension, which is then interrogated with either BD Precise plates or the Resolve system.”
Researchers sort single cells of desired phenotypes into specific wells of a Precise 96-well plate, enabling correlation of protein and RNA expression of the same cell, explains Dr. Shum, adding that with the Resolve system, up to 10,000 sorted cells can be simultaneously interrogated. Both options provide methods for defining cell-to-cell variation, either within a cell population or across different cell types.
“With both Precise and Resolve, single cells are first lysed, then each mRNA molecule is barcoded during cDNA synthesis,” according to Dr. Shum. The resulting cDNA is pooled into a single tube for final library creation.
“This technology fits perfectly for single-cell mRNA sequencing of hundreds to thousand cells with a simplified workflow and resolution of single cells at an affordable price,” she says. “The power of a streamlined workflow from single-cell preparation to targeted or whole transcriptome analysis provides a complete solution for high-throughput single-cell gene expression.”

