Until very recently, adeno-associated virus (AAV) had an excellent safety record as a viral vector for gene therapies, which goes a long way to explain its widespread use in this capacity. However, the question of immunogenicity and particle toxicity—especially at high doses of AAV—is rearing its ugly head.
Some immune responses to AAV, such as the development of capsid-specific neutralizing antibodies or effector T-cell responses, have been well studied. Others, such as incidences of neuroinflammation or thrombocytopenia that have sometimes been observed following high-dose treatments, are less well understood.1 We also don’t know whether improving methods of AAV manufacturing, such that we could reduce or eliminate the number of empty capsids, improve preparation purity, or lower the minimum effective dose of AAV, might help counter some of these responses and improve the safety profile of this important viral vector further.
Over the last three years, one of our teams at OXGENE has had a single mission—fundamentally change AAV manufacturing. We have explored many approaches, including stable “producer” cell lines, plasmids with improved gene configurations, semi-transient processes, and “packaging” cells that supply key components in trans, but none of these approaches delivered the step change we were looking for.
Reassessing the prevailing method
In nature, AAV shares an exquisite relationship with adenovirus. Relying on adenovirus allows AAV to gain most of the functionality it needs for its life cycle. This highly evolved and dynamic relationship results in exceptionally high-fidelity production of AAV, with almost every AAV particle receiving a correct genome. As such, for the recombinant manufacturing of AAV, where the rep and cap genes have been removed, “helper” adenovirus has been used extensively as a manufacturing tool. However, the unpurified AAV prep frequently contains as much helper adenovirus as it contains AAV. The removal of this adenovirus represents a major challenge for downstream processing and a significant safety issue.
For these reasons, over the past decade, recombinant plasmids encoding just the essential adenovirus and AAV components have prevailed as the main method of AAV manufacturing. Although this “helper free” (or “adenovirus helper free”) method provides a safer and cleaner approach, the trade-off for this is that the system is not easily scalable. This is an increasingly limiting factor as our understanding of the genetic basis of disease increases, and as the industry moves toward the development of gene therapies for systemic diseases or diseases with large patient populations. Scalable, high-quality manufacturing of AAV will be key to the success of these endeavors.
Realizing a new concept
Our team started with the conceptual premise that replicating nature by using adenovirus to make AAV would be the best approach. We knew that contamination of the AAV prep with adenovirus particles would be the major challenge to resolve, and we eventually cracked this by manipulating the adenovirus life cycle.
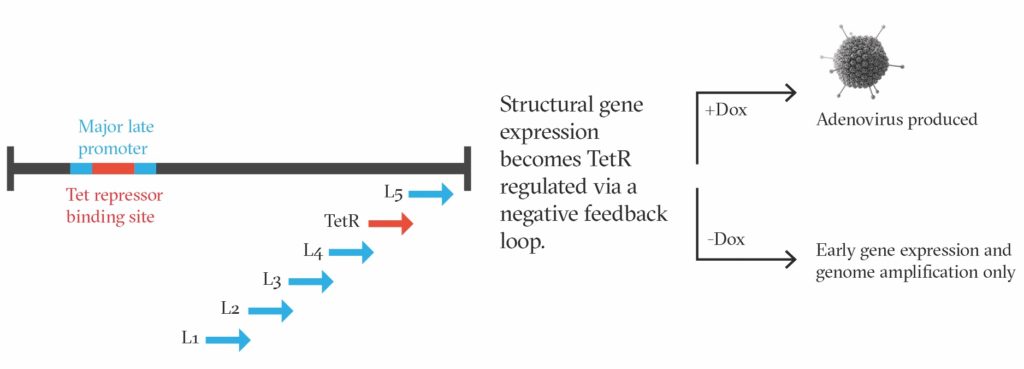
Conveniently, adenovirus has two intracellular life cycle phases, early and late. Although the early phase is crucial for AAV production, the late phase is not required at all. Rather handily, the late phase is wholly responsible for making the adenoviral structural components and is therefore the source of all unwanted adenovirus contamination.
We set out to find a way to turn the late phase of the adenovirus life cycle off when making AAV, and only switch it back on when we wanted to produce more of the helper adenovirus itself (as a reagent). This would allow us to use adenovirus as a helper for efficient manufacturing of AAV without any concomitant adenovirus contamination, since the cells would be unable to make adenoviral structural proteins and should, consequently, produce only pure AAV in the end.
To ensure supremely tight regulation of the adenovirus late phase, we built in a proprietary negative feedback loop, and we have demonstrated that this can repress adenovirus production by 99.999999–100% during an AAV manufacturing run. We call this TESSA—a Tet-enabled self-silencing adenovirus. When we need to produce more TESSA vectors, we simply add doxycycline to turn the late phase of the life cycle back on, and we grow the vectors to high titer in HEK293 cells (Figure 1).
As a first step, we demonstrated that TESSA technology could improve the quality of AAV preparations when it was introduced alongside two plasmids—one containing the recombinant AAV genome, and the other encoding the AAV genes rep and cap. While this demonstrated that the TESSA system worked, it didn’t make the system any more scalable, as it still required plasmid transfection. So, we took it a step further and attempted to insert the rep and cap genes into the TESSA adenovirus to reduce the number of plasmids required.
After many failures, reconfigurations, and optimizations, we have now produced stable TESSA vectors that contain both the AAV rep gene and the AAV cap gene. This is the first time that both rep and cap have been stably included within an adenoviral vector, a vector that allows the efficient simultaneous delivery of all the adenovirus helper functions, alongside all the AAV components in a single agent, without any transfection. We have so far successfully demonstrated this for AAV2, AAV5, AAV6, and AAV9.
Providing the AAV genome, or not
The only component that still needs to be provided separately is the AAV genome, so we considered how best to achieve this. We tested plasmid transfection, infection with a second TESSA vector encoding the AAV genome (which gives more efficient delivery than transfection), and a cell line with an integrated AAV genome. Of the three, the second TESSA vector provided the best results.
Overall, for AAV2, the TESSA system gives approximately a 20-fold increase in AAV particle yields compared to the helper-free system. Additionally, we see a >2,000-fold cumulative increase in AAV2 infectious yield and increased the percentage of full capsids from 2–5% up to 70% (Figure 2A).
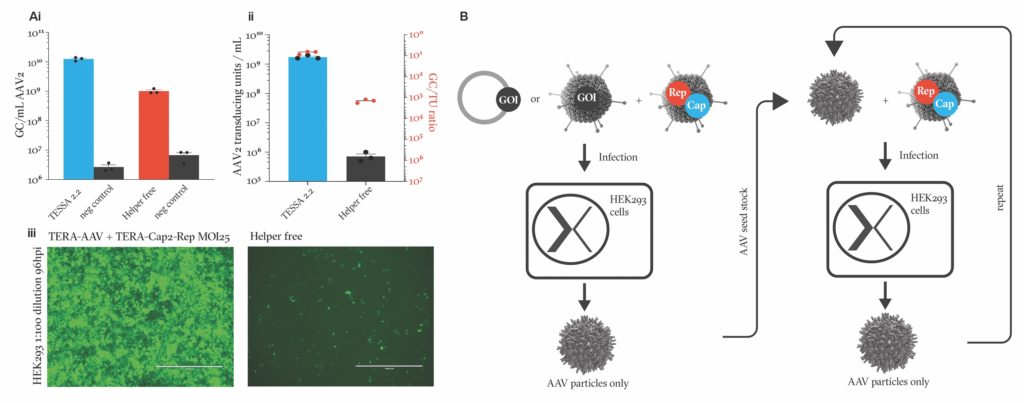
This was the point at which we realized we’d missed a trick. We’d been worrying about the best and most scalable way to provide the AAV genome. But we didn’t need to. Because once we’d made and harvested the first rAAV preparation, we could infect cells simultaneously with both this rAAV and more TESSA vector, and it would propagate our rAAV seed stock still further. Theoretically, you could repeat this step, producing more and more pure AAV in a simple, easily scalable, and contaminant-, plasmid-, chemical-, and transfection-free process (Figure 2B).
Conclusion
TESSA technology represents a radical—and much needed— overhaul of AAV manufacturing. It is completely scalable and significantly reduces the number of input materials required. If you already have GMP-grade rAAV, you can simply grow more of it. If you only have a plasmid with your AAV genome in it, you can recover a small amount of rAAV and then grow it repeatedly, without ever needing any more plasmid.
We hope that this technology will forever change the way AAV is manufactured, resulting in reduced costs, improved quality, and a better safety profile of a class of medications that patients desperately need.
Ryan Cawood, DPhil, is CEO of OXGENE.
References
- Flotte TR. Revisiting the “New” Inflammatory Toxicities of Adeno-Associated Virus Vectors. Hum. Gene Ther. 2020; 31(7–8):398–399. http://doi.org/10.1089/hum.2020.29117.trf.
