
There is a certain irony that the first approved CRISPR therapy—a technology barely 10 years old—should be for the genetic disorder that Linus Pauling famously dubbed “the first molecular disease” almost 75 years ago. On December 8, 2023, the U.S. Food and Drug Administration (FDA) approved Casgevy, a groundbreaking CRISPR-based gene editing therapy from Vertex Pharmaceuticals and CRISPR Therapeutics, for sickle cell disease (SCD). The approval was never seriously in doubt, as the therapy—also known as exagamglogene autotemcel, or exa-cel—demonstrated spectacular clinical results dating back to the first patient, Victoria Gray, who was dosed in July 2019.
The FDA also announced approval of another SCD gene therapy, Bluebird Bio’s lentiviral therapy, Lyfgenia. However, that approval comes with a black box warning given the occurrence of rare instances of blood cancers in patients.
“Going from the lab to an approved [CRISPR] therapy in just 11 years is a truly remarkable achievement,” said Nobel laureate Jennifer Doudna, PhD, following news of the United Kingdom’s approval of Casgevy last November. She was especially pleased because the approval helps patients with “a disease that has long been neglected by the medical establishment.”
[Fyodor Urnov/The CRISPR Journal]
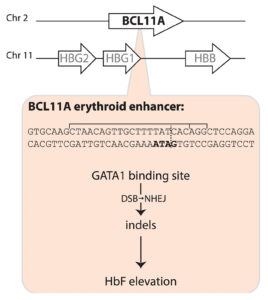
The Casgevy strategy is to compensate for the SCD mutation in the b-globin gene by restoring expression of fetal hemoglobin (HbF), which is expressed in utero and switched off shortly after birth. The CRISPR-Cas9 scissors is applied in patients’ harvested stem cells to inactivate the BCL11A repressor, which in turn releases the handbrake on HbF expression.
That target was identified some 15 years ago, in foundational genome-wide association studies led by Vijay Sankaran, MD, PhD, Lodish Family Chair in the Division of Hematology/Oncology at Boston Children’s Hospital, and independently by Swee Lay Thein, MD, chief of the sickle cell branch at the National Institutes of Health (NIH).
“This experience underscores a critical lesson—the indispensable role of fundamental discovery science,” Sankaran told GEN. “Without such studies, many of these pivotal advances would remain beyond our reach.” Thein agreed, telling GEN, “This is a major triumph for translational science and a major step toward curative treatment for patients with sickle cell disease. … But let’s not forget the rest of the patients; we still need more small-molecule disease-modifying drugs for patients with sickle cell disease.”
Among other notable reactions to the FDA news was Victoria Gray. “I am crying real tears of joy, and I can’t stop shaking!!,” she posted on LinkedIn. “To all my fellow Sickle Cell Warriors help is here! We will never forget the warriors who we lost along the way! … Our prayers were not in vain! This is only the beginning!!!”
In Tanzania, Julie Makani, MD, PhD, a leading SCD physician-scientist, hailed the news as “a momentous milestone” for SCD, one that would have “a major impact on our approach” to SCD therapeutic interventions. Two decades ago, “the message to patients was that there is no cure for SCD,” she told GEN. “Today, we can tell patients that gene therapy is available for treatment of SCD in the USA and UK.” Makani is leading efforts to expand access to gene therapy for SCD patients in Africa, which is where the SCD mutation first arose more than 7,000 years ago.
A century of hurt
Sickle cell disease affects some 100,000 people in the United States, the vast majority African Americans, and millions more worldwide. A point mutation in the b-globin gene results in the polymerization of the oxygen-carrying hemoglobin protein. That in turn warps the shape of the beautiful, biconcave red blood cells, leading to blocked blood vessels, excruciating pain crises, and the risk of organ damage.
In 1910, Chicago physician James Herrick, MD, published a case report of an unnamed 20-year-old dental student from the Caribbean island of Grenada, who had first been treated in 1904 by a resident, Ernest Irons, MD. Herrick noted the presence of peculiar “pear shaped” red blood cells. Following a few additional case reports over the next decade, Johns Hopkins resident Verne Mason, MD, coined the term “sickle cell anemia” in 1922.
Herrick’s patient was identified decades later by historian Todd Savitt, PhD, as Walter Clement Noel, who arrived in Chicago in 1904 to enroll in dental school. He returned to Grenada to set up his own dental practice in 1907 but died less than a decade later in 1916. Noel is buried in a family grave on Leapers Hill, a famous landmark on the north coast of the island, in a Catholic church cemetery overlooking the ocean.

Two major discoveries occurred in the late 1940s. In 1948, a Long Island pediatrician named Janet Watson, MD, deduced that higher levels of HbF, such as in SCD patients shortly after birth, was correlated with a reduced level of sickling—an important observation that laid the foundation for the CRISPR strategy employed by Casgevy. A year later, Linus Pauling, PhD, demonstrated that SCD was a recessively inherited disease caused by a molecular alteration in hemoglobin.
Seven years later, Vernon Ingram, PhD, working in the same Cavendish Laboratory where Crick and Watson assembled the double helix model of DNA, identified the precise amino acid substitution in b-globin. Around the same time, epidemiological studies in East Africa by Anthony Allison, MD, proved that the incidence of SCD coincided with rates of malaria—a vivid example of heterozygous advantage.
Rewriting the medical system
But despite these and other scientific milestones, the experience of SCD patients has not advanced to the same degree. As the late science journalist Sharon Begley put it: “The U.S. healthcare system is killing adults with sickle cell disease.” And while FDA approval of Casgevy is a moment for celebration, this approach won’t help the vast majority of patients worldwide. As Dhruv Khullar wrote in the New Yorker: “If we truly want to cure sickle cell disease, editing genomes will only get us so far. We’ll need to rewrite our medical system, too.”
Even today, more than a century after the first case report, SCD patients are still subject to profiling and suspicion by hospital staff as they seek pain medications. “There may be no population of patients whose healthcare and outcomes are more affected by racism” than those with SCD, hematologists Alexandra Power-Hays, MD, and Patrick McGann, MD, wrote in the New England Journal of Medicine. Making matters worse, many SCD patients lack basic information and/or access to generic drugs and screening tools that could ward off disease complications.
As the gene and cell therapy field celebrates another approval for precision medicine, we should be ashamed that fewer than one in five children with SCD are prescribed antibiotics or the generic anticancer drug hydroxyurea, which boosts levels of HbF. “To have teenage patients who never heard the word hydroxyurea—that’s preposterous,” McGann declares. There isn’t even a national registry of SCD patients, so no one can say precisely how many individuals are affected.
More than 50 years ago, President Nixon signed the Sickle Cell Control Act, creating new treatment centers and increased funding. But the benefit was short lived. In a study published in 2020, Duke University hematologist John J. Strouse, MD, and colleagues argued that by many metrics, including federal funding, philanthropic support, and new drug approvals, support for SCD lags that afforded cystic fibrosis, even though cystic fibrosis affects roughly one third of the number of patients in the United States as SCD.
A new hope
In August 2022, Pfizer acquired Bay Area biotech Global Blood Therapeutics (GBT) for $5.4 billion. GBT’s small-molecule drug Oxbryta binds to the b-globin molecule and prevents polymerization. Other small-molecule drugs are showing promise in the clinic.
But the value of gene and cell therapy approaches is that they promise “one and done” therapies that do not depend on a relative to donate stem cells. NIH hematologist John Tisdale, MD, who has been treating SCD patients with Bluebird Bio’s lovo-cel, a form of gene therapy that uses a lentiviral vector, was profiled on 60 Minutes two years ago. The program included a comment from former NIH director Francis Collins. He said, “This looks like a cure.”
Meanwhile, Beam Therapeutics and Editas Medicine are also pursuing SCD among their lead gene editing programs. At Boston Children’s Hospital, David Williams, MD, and Erica Esrick, MD, have published promising clinical data treating SCD patients using a short hairpin RNA approach, again targeting the BCL11A pathway.
Taking over the exa-cel program launched by CRISPR Therapeutics, Vertex published the initial exa-cel results in the New England Journal of Medicine in 2021. These represented not only a huge advance in treating SCD but also a crucial early validation of the clinical promise of CRISPR. Fyodor Urnov, PhD, scientific director at the Innovative Genomics Institute, hailed the results as “borderline utopian.”
Writing in The CRISPR Journal, Urnov proposed that Victoria Gray—who has been featured in a series of interviews on National Public Radio since 2019—be added to “the pantheon of names inscribed in golden letters in the history of biomedicine.” That list includes James Phipps (the boy vaccinated by Edward Jenner), Albert Alexander (the first recipient of penicillin), Louise Brown (the first test tube baby) and Emily Whitehead (the pioneering chimeric antigen receptor T-cell patient).
Utopian or not, the positive exa-cel trial results have been extended to dozens of SCD patients, with no reported serious adverse events, although a couple of patients have continued to experience some vaso-occlusive events. The only potential concern among FDA regulators was the possibility of CRISPR-induced off-target effects.

Vertex Pharmaceuticals
Five weeks ago, an FDA advisory committee meeting assembled a panel of experts to consider this issue. The Vertex investigators, led by chief science officer David Altshuler, MD, PhD, satisfied most of the panel’s lingering concerns about off-target editing and emphasized plans to monitor the long-term health of the exa-cel trial volunteers.
The FDA’s concern is understandable. In August 2022, Graphite Bio, a Bay Area gene editing company co-founded by Stanford University physician-scientist Matthew Porteus, MD, PhD, launched its own SCD gene editing trial. But five months later, the company voluntarily paused the trial after its first patient developed complications. Graphite Bio subsequently halted the trial completely, downsized, and agreed to a reverse merger with LENZ Therapeutics, a biotech company developing ocular therapies. Porteus has since launched a new company, Kamau Therapeutics, and acquired the rights to the SCD program, believing that the adverse events suffered by the first patient were caused by a drug used to treat low platelet counts and unrelated to gene editing.
Long road ahead
Immediately following Casgevy’s approval, Vertex announced a list price of $2.2 million, while Bluebird priced Lyfgenia at $3.1 million. But an ex vivo genome editing protocol that involves toxic chemotherapy, stem cell harvesting, and weeks in hospital is not going to be easily affordable or scalable.
If Gray’s inspiring story is the medical equivalent of the Lindbergh flight across the Atlantic, the next challenge, Urnov says, is to develop the equivalent of “routine, scalable, safe, and reasonably priced affordable air travel in a Dreamliner.” Urnov has been advocating regulatory and organizational incentives to fully realize the clinical potential of gene editing (see his recent video interview on GEN’s “Close to the Edge”).
How will these therapeutic advances be translated to the millions of SCD patients living in Africa, Asia, and beyond? Speaking at GEN’s State of Biotech virtual event in 2022, Doudna predicted that a one-time in vivo delivery approach—without ex vivo manipulation and bone marrow transplantation—would ultimately be achievable. “Is that going to be possible? My answer is yes,” she said. “Is it possible today? No.”
For SCD patients suffering from “the most famous point mutation in genetics,” as Beam’s CEO John Evans calls it, Casgevy is not the end of the journey, merely a long-awaited beginning.
