
In biosafety testing, the expression “dot your i’s and cross your t’s” comes to mind. If only biosafety testing were so simple. But it isn’t. Yes, biosafety testing does require that we attend to the details of well-established tasks, such as ensuring the quality of therapeutic antibodies and recombinant proteins. But biosafety testing must also deal with tasks that are relatively new and less well defined, tasks involving the production of gene therapies and cell-based therapies. It is almost like working with an alphabet that keeps adding new letters.
Biosafety testing must be both meticulous and innovative given the emergence of biopharmaceutical products such as recombinant adeno-associated virus (rAAV) vectors. New kinds of products come with new challenges, making it necessary to consider new solutions.
Biosafety challenges and potential solutions were discussed at two recent meetings. Valuable insights from these meetings are presented in this article, which includes commentary from scientists who spoke at the Bioprocess International Conference (which was held last September) or the Bioprocess Summit (which was held last August).
The scientists quoted in this article address a range of topics: robust viral inactivation and/or removal technologies that can be applied to diverse vector manufacturing processes; applications of two-dimensional HPLC (2D-HPLC) that can improve the monitoring of process yield and critical quality attributes; quality-by-design methodologies that can help developers overcome shortcomings in Chemistry Manufacturing and Controls (CMC) documentation; the use of orthogonal approaches in host cell protein detection; and the use of cell-free cloning technology to avoid the complications associated with ill-defined cell lysates.
Biosafety strategies for rAAV vectors
For therapeutic antibodies and recombinant proteins, biosafety profiles are fairly straightforward. The same cannot be said for rAAV vectors. “The production systems, cell substrates, and recovery technologies for biologics have been well investigated for more than two decades,” notes Shengjiang (Shawn) Liu, PhD, the president and CEO of Avirmax, a company that is pursuing AAV-vector-mediated biotherapeutics based on Bac-to-AAV technologies. “A lot of process data and knowledge have been accumulated for therapeutic antibodies and recombinant proteins.”
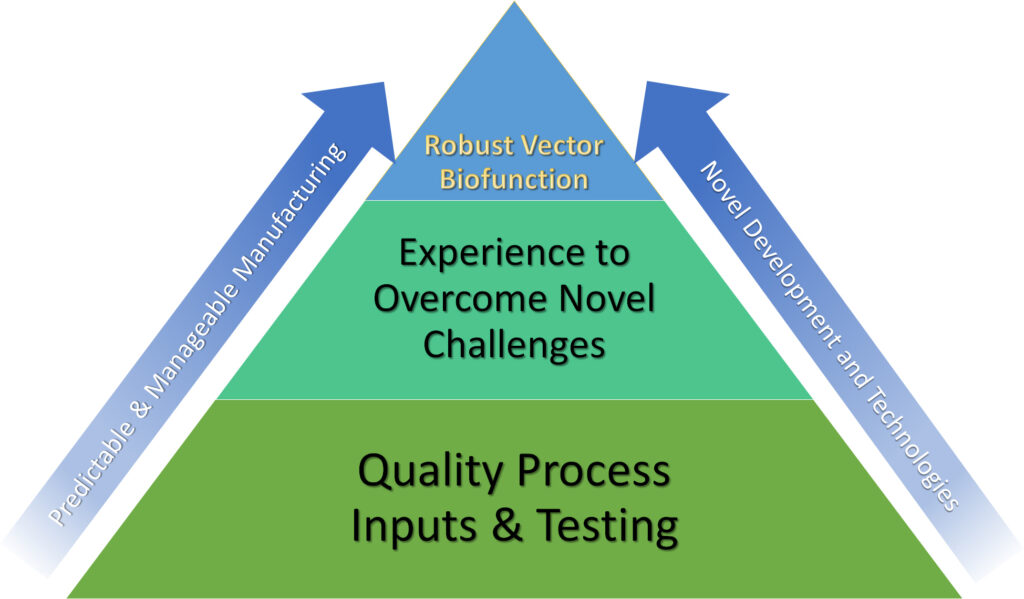
“The biosafety profiles for these biologics are more predictable and manageable,” he continues. “However, achieving a full biosafety profile for gene therapy vector production is in its infancy [in terms of] the technologies and accumulated experience [that will be] necessary.”
Liu says that biosafety approaches will vary with vector biofunction: “Enveloped viral vectors (for example, lentiviral vectors) and non-enveloped vectors (such as rAAV vectors) are dramatically different from each other, and these vector types also have very different biophysical and biochemical properties. Therefore, the strategies and methodologies used in gaining biosafety should be considered differently.”
He elaborates, “One of the biggest challenges is to develop suitable, effective, and robust viral inactivation or removal technologies applicable to the diverse vector manufacturing processes.” For example, companies must mitigate the risks of potential transmission of pathogens such as endogenous virus-like particles, adventitious agents, and/or transmissible spongiform encephalopathies that may result from using the human-derived cell substrates in vector production.
Liu is optimistic that the industry will solve its challenges: “Since the first recombinant protein that was approved for human use, there has been no single case of viral infection/transmission caused by a therapeutic product manufactured using recombinant technology. I believe this will be true for gene therapy vector production.”
2D HPLC for early-stage development
Monitoring process yield and critical product quality attributes during the downstream processing of AAV-based drugs can provide helpful safety information. “The amount of empty capsid in an AAV drug substance can impact product efficacy and trigger immune responses,” says Xiaotong Fu, PhD, senior engineer in Biogen’s gene therapeutics process development team. “Monitoring the percent of empty capsid during process development allows us to manufacture drug substance with consistent purity and a consistent safety profile.”
Fu reports that Biogen is focused on a 2D-HPLC approach because it offers several advantages: “The 2D-HPLC approach … is widely accepted by quality control teams and regulatory agencies. It’s automated and has higher throughput, whereas other methods may struggle with manual operation or low throughput. Also, it consumes less material, making AAV sample analysis more cost efficient, allowing more extensive testing if needed.”
Biogen’s 2D-HPLC method uses a two-column process to purify AAV from crude samples. It takes ultraviolet absorbance measurements of purified AAV and converts them to titer and empty/full information. “This method agrees well with other orthogonal methods that are established in the gene therapy field,” Fu asserts. “It provides us an option to track AAV titer and empty/full percentage in a high-throughput, automated, and cost-efficient way. It means we can better assess product quality, including biosafety, from early-stage development.
Fu also indicates the 2D-HPLC method enables process monitoring both for upstream and downstream processes via a rapid, at-line measurement without exhausting purification and analytical support resources. Biogen continues to refine this method for better accuracy and robustness.
For the future, Fu feels the industry needs more novel approaches to address biosafety issues. “Right now, options to characterize AAV product biosafety profiles are still limited, especially for those options that can speed up this characterization process and improve cost-efficiency,” he points out. “I’d encourage the field to keep working on innovative approaches for gene therapy products, which will benefit the gene therapy field, as well as patients, in the long run.”
An ounce of QbD
In 2020, the FDA rejected biologics license applications for several gene therapy products because they offered too little chemistry, manufacturing, and controls (CMC) information. “Poorly prepared CMCs,” observes Parth Trivedi, team manager, Pall, “represent one of the largest obstacles for companies moving toward regulatory approval for new gene therapy drug candidates.”
Trivedi and his team members advise that a key aspect of appropriate CMC documentation is the application of quality by design (QbD) principles. “QbD relies on process design rather than final quality testing alone and focuses on achieving safety and efficacy from the very beginning,” Trivedi emphasizes. “Usually, this type of information is heavily based on prior knowledge. However, for gene therapy products, experience is still very limited, making it difficult for manufacturers to draw on accumulated knowledge.”
To address this knowledge gap, Pall provides a template that is especially designed for AAV platforms. Trivedi explains, “The QbD approach begins by defining a quality target production profile that summarizes specifications for a drug product’s safety, purity, and efficacy. This analysis will identify and emphasize critical quality attributes (CQAs). To assure that these CQAs are met, two more processes are required: the identification of a set of critical process parameters, and the definition of critical material attributes.”
Trivedi notes that Pall has prepared a white paper (“QbD for AAV”) that describes an QbD-based AAV reference process framework and applies it to the four most common upstream production methods: 1) transfection of adherent human embryonic kidney (HEK) cells; 2) transfection of suspension HEK cells; 3) baculovirus infection of Sf9 suspension cells; and 4) adenovirus infection of HeLa producer cells.
“We focused on the effect on patient safety and identified 14 CQAs for the AAV platforms,” Trivedi reports. “We combined literature, industry, and in-house data as well as knowledge from subject matter experts at our company.”
According to Trivedi, the take-home message is straightforward: “We’ve shown that a wide process understanding has already been created within the industry, despite the newness of AAV manufacturing for biopharmaceutical production. Going forward, the best advice is to consult with experts in the field.”
New angles on HCPs
Residual host cell proteins (HCPs)—proteins that will, inevitably, contaminate biopharmaceutical products—represent a dimension of CQA biosafety monitoring in the manufacture of gene therapy products. AAV vectors are often manufactured in human cells lines such as HEK293. This approach leads to a complex proteomic background. Additionally, harvesting viral vectors involves cell lysis, potentially contributing to increased HCP loading.

Charles River
Andrew Hanneman, PhD, scientific advisor, Biologics Testing, Charles River Laboratories, says that the limited number of downstream purification steps in viral vector production may lessen the capacity to separate vectors from background HCP impurities. This problem may be exacerbated in gene therapy programs, which are intended for severe indications and are thus more likely to be run at an accelerated pace. When faced with time constraints, companies may choose to forgo product-specific immunoassays during biosafety analysis. That is, the time needed to develop such assays may seem too dear. An alternative approach to product safety, Hanneman suggests, is to rely on mass spectrometry.
“Traditional product-specific ELISA development using a process-matched host cell protein antigen (null or mock material) to support Phase III clinical lot production is time consuming and requires a significant amount of material,” he points out. “AAV companies are, therefore, often relying on generic HCP kits. However, ELISA kits may not provide reliable HCP coverage to detect all risk proteins. Relying on kit-based
ELISA results without supporting data to understand the kit’s HCP coverage represents a safety and compliance risk.”
The solution Hanneman proposes is to identify and quantify HCPs using liquid chromatography with tandem mass spectrometry (LC-MS/MS) implementing a label-free quantification (LFQ) workflow. He indicates this methodology represents an unbiased orthogonal approach to ELISA, one that also provides a means of identifying individual protein contaminants. He adds, “A related MS-based quantification method for any high-risk proteins identified can use peptide standards which are spiked into samples at defined concentrations to provide an absolute quantitative readout of target HCP content.”
These approaches can also be coupled with ELISA coverage methods. He elaborates, “ELISA kit coverage by traditional 2D gel western blot is useful, as are newer approaches using immunoprecipitation together with LC-MS/MS to determine which proteins are or are not recognized by the ELISA reagents.” In these approaches, kit-based or process-specific reagents may be used.
A debugged cloning process
Regardless of the gene therapy platform employed, the input of raw materials weighs heavily on subsequent biosafety. One new strategy, according to Nasir Bashiruddin, PhD, chief technology officer, OriCiro Genomics, is to utilize a cell-free cloning technology. One such technology has been developed by OriCiro. “Since our technology does not require cells, the requirement for antibiotics and their corresponding resistance genes are not needed for the manufacturing process,” he asserts. “Further, our Cell-Free Cloning System is fully reconstituted from purified proteins and therefore contains no endotoxin or host cell DNA and RNA.”
Bashiruddin suggests that a cell-free system has many advantages: “Plasmid DNA manufacturing with our system is highly scalable, so moving from early studies to GMP level will require much less lead time yet not require the creation of master cell banks. Being able to circumvent the need for master cell banks also removes concerns over plasmid loss, copy number, and supercoiled DNA content. Once the plasmid DNA is synthesized, the downstream purification is very simple and fast due to it being a fully defined system, unlike cell lysate. Finally, our major synthesis product is supercoiled DNA, which is the most desirable form for downstream manufacturing.”
As companies make strides to advance gene therapy products, parallel progress in rigorous biosafety testing must also be achieved emphasizing the need for out-of-the box thinking, as well as nontraditional uses of traditional technologies.