
Making a therapeutic that contains biological components is just the start of creating a medicine. Then, biopharmaceutical companies must capture that drug and ensure that it’s free of contamination. New techniques and technologies can accomplish that capture and cleanup more simply and economically.
New approaches to downstream processes dominated the agenda at the fifth annual Advances in Purification & Recovery, one of the conferences making up Cambridge Healthtech Institute’s Bioprocessing Summit, which was held August 14–15 in Boston. The conference’s speakers emphasized the intensification and optimization of downstream processing. We asked five of the speakers, downstream processing’s leading scientists, to share their insights with our readers.
Achieving custom capture
Monoclonal antibodies (mAbs) constitute the most successful biologics category, but they account for a minority of the biologics currently in development. “They represent only 35 to 40% of all biopharmaceutical drug modalities in registered pipelines,” said Karol Lacki, PhD, vice president of technology development at Avitide, “and other types of biological modalities are gaining more attention.”
Some of the non-mAb modalities create manufacturing challenges, and companies are working to develop purification processes for these modalities that work as robustly as those for mAbs. “If the first step in your purification process is very specific—for example, if you use protein A to purifiy mAbs—the whole process is simplified,” Lacki explained. Plus, a lower number of purification steps produces a higher overall process yield, which increases production rates and lowers cost.
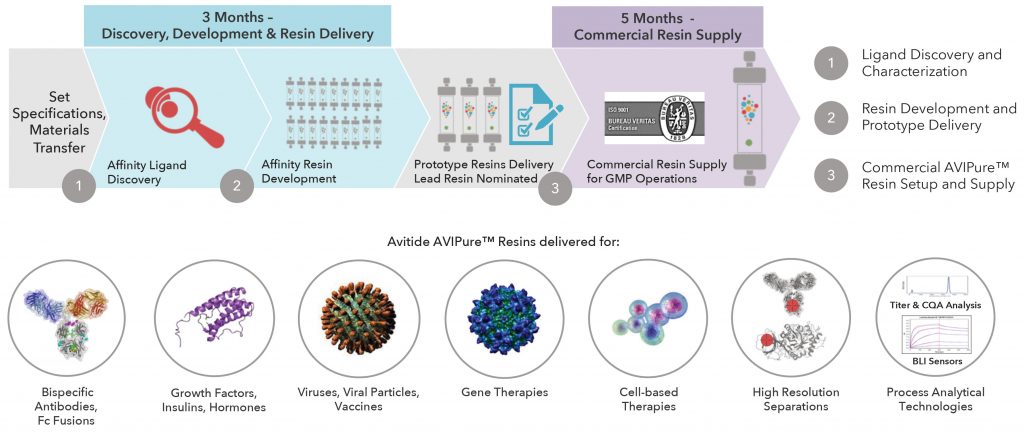
Avitide develops custom affinity resins for standard capture applications and resins to solve extremely difficult purification challenges, where closely related product impurities need to be separated from the desired product. As examples, Lacki mentions Avitide’s resins for separating oxidized and nonoxidized versions of the same molecule, folded versus unfolded protein therapeutics, full-length versus truncated, and active versus inactive forms of a therapeutic enzyme.
Avitide develops resins in three months. “The resin prototypes developed after the 12 weeks are evaluated by the customer for future implementation in a larger scale process,” Lacki noted. “We can produce GMP-ready resins, within our industry-certified quality management system, in four to five months with volumes of resin that fulfill the customer’s needs.”
Custom affinity resins allow a company that produces mAb-based therapeutics to leverage the same facility and equipment to manufacture non-mAb products. “You just use a different resin in the same column and potentially a different buffer as compared to your mAb process,” Lacki pointed out. “That gives people more flexibility in deciding where they will manufacture their non-mAb biologics.”
Making use of magnetic nanoparticles
Increasingly diverse and abundant diagnostic and therapeutic proteins can enable new medical approaches, but much depends on economic considerations. Fortunately, cost reductions and productivity gains may be achieved if production processes are improved.
“For the separation of proteins, magnetic biocompatible adsorbers offer a new alternative to classical solid-bed chromatography methods of downstream processing,” said Sonja Berensmeier, PhD, professor of bioseparation engineering at the Technical University of Munich. “If the magnetic particles have a high selectivity, like affinity-chromatographic material, they can be used directly in the capture step and fish out the target molecule directly from a broth containing solids.”
Beyond the material used, the size of the particles also matters. “If nanoparticles are used instead of microparticles, which are already state of the art in analytics, a much higher binding capacity can be achieved than in classical chromatography,” Berensmeier continued. “A further advantage of such nanoscale particles is that they are nonporous, and therefore, no mass transfer limitations occur.”
When biocompatible nanoparticles incorporate an iron oxide matrix, they are superparamagnetic. “They can be magnetized by a magnetic field, but [they] lose their magnetic properties again as soon as the field is switched off,” Berensmeier explained. That’s a perfect property for processing samples. Overall, these nanoparticles are easy to produce and functionalize.
So far, high-gradient magnetic separators from the water treatment industry have been modified for technical separation. “In the meantime, the first separators have been developed that also meet the hygienic requirements of the pharmaceutical industry and can separate nanoparticulate iron particles from larger volume flows,” Berensmeier stated. “Preliminary work on the separation of tagged proteins and antibodies shows that the technology has a high potential and represents an alternative to cost-intensive affinity chromatography.”
Spiking processes with noninfective particles
Biopharmaceutical manufacturing processes are prone to viral contamination. Consequently, these processes, which include clinical and commercial processes, do not receive regulatory approval until their viral clearance capabilities are confirmed. The U.S. Food and Drug Administration (FDA) and international regulatory agencies usually require a viral clearance spiking study. In such a study, a virus is added to test runs of the process to quantify virus removal or inactivation.
Viral clearance spiking studies are typically outsourced to specialized contract research labs. These studies are expensive, and most companies delay them studies until a biologics license application (BLA) is submitted. “The cost of performing a viral clearance study is the number one challenge that limits a company’s ability to explore the potential for viral clearance during process development and characterization,” said David Cetlin, CEO, MockV Solutions.
So, scientists at MockV Solutions developed a mock virus particle (MVP) based on a parvovirus—minute virus of mice (MVM)—typically used in validation studies. The MVP consists of the “proteinaceous shell of the real virus without the infectious genetic elements, such as nucleic acid,” Cetlin explained. “Then, a customer would challenge their process with our noninfectious particles and use our quantification reagents to analyze the viral clearance.”
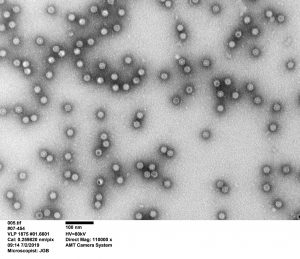
MockV Solutions’ MVM-MVP kit costs $4,000, and it can be used to run about 10 small-scale experiments. “The typical viral clearance study costs tens if not hundreds of thousands of dollars,” Cetlin pointed out.
Although the current kit does not match the sensitivity of a live virus infectivity or quantitative polymerase chain reaction (qPCR) assay, Cetlin’s team keeps improving it. Plus, the current kit provides useful information.
As an example, U.K.-based GlaxoSmithKline (GSK) approached MockV Solutions to run a design-of-experiment study with the MVM-MVP kit. “GSK wanted to see what would happen if it ran an anionic-exchange chromatography column at a different pH or conductivity,” Cetlin recalled. In just two weeks and one kit, GSK used the results to build a model of the impact of the changes. “With traditional technology,” Cetlin maintained, “that might have taken a month and at least $30,000 to $40,000.”
Quantifying impurities in cell therapies
To know what a biomanufacturing process fluid must be rid of, scientists must know the fluid’s contents. Only then can scientists efficiently purify the fluid, yielding an acceptable biotherapeutic.
New ways of being in the know are being pursued by Hai Yue, PhD, senior scientist, analytical development, Juno Therapeutics, a Celgene company. His task is to characterize and understand potential impurities in cell-based therapeutics, specifically, chimeric antigen receptor (CAR) T cells for treating B-cell malignancies.
Yue and colleagues develop CAR T-cell therapies from a patient’s white blood cells. These therapies are derived from samples that can include unwanted cells or process-related impurities, such as components of cell media or materials related to gene editing, such as a residual virus vector. The goal for Yue and colleagues is to understand the impurity profile of a CAR T-cell therapy.
The diverse collection of cellular drug products at Juno/Celgene requires a collection of analytical tools to detect impurities. The tool used “depends on the impurity we’re looking at,” Yue noted. To quantify unwanted cellular impurity, for example, Yue uses flow cytometry. He measures residual nucleic acids with PCR. “To provide more in-depth understanding,” he continued, “we use orthogonal methods.”
Building multistep processes
Other groups also explore new approaches to testing biotherapeutics. At Texcell North America, director of research and development Akunna Iheanacho, PhD, and her colleagues are part of a global research contract organization that provides viral clearance services and viral safety testing in support of applications submitted to the FDA and other regulatory authorities.
“At Texcell,” Iheanacho said, “we have cultivated a range of experience performing viral clearance studies for a variety of biopharmaceutical products, including mAbs and gene therapies as well as medical devices.”
A robust viral clearance program depends on understanding the manufacturing process used to develop the biological product. “In a nutshell,” Iheanacho explained, “you are testing the capacity of the manufacturing process to clear a broad spectrum of viruses.”
Scientists use different strategies for removing and inactivating viral contamination. “For virus removal, a typical approach is to use chromatography, virus filtration, or both, while the use of heat, irradiation, solvents, detergents, or low pH are all treatment techniques employed to inactivate virus,” Iheanacho pointed out. “Other considerations include the selection of an appropriate virus panel that includes both enveloped and non-enveloped viruses.”
The European Medicines Agency (EMA) regulatory guidance asserts that reductions of 4 log10 or more indicate a clear effect with the virus under investigation, while process steps with 1 log10 or less of clearance are considered negligible. “Relying on one process step to clear all adventitious viruses is not the best strategy,” Iheanacho cautioned. “For instance, if you utilize an anion exchange chromatography step and viral filtration step for virus removal that each yield a log reduction value of 5 or greater, coupled with a low pH hold step for virus inactivation with a log reduction value of 6 or greater, then you’re setting up for success, because each of these process steps employs a different mechanism of action and contributes to the overall virus-reduction potential.”
As biology-based therapeutics get more complex, drug makers need more advanced tools and techniques to create safe products and economical processes. Undoubtedly, new tools—such as capture resins, test kits, and more—will continually improve purification and analytical capabilities, but new methods also matter, such as combining the right series of downstream steps.
