
October 15, 2008 (Vol. 28, No. 18)
Tips on Choosing the Best Assay and Optimizing It Effectively
The last two decades have witnessed the steady growth of the protein drug market. As original recombinant products come off patent, generics/ biosimilars are also beginning to be manufactured.
The increase in the number of protein drugs has led to a greater focus on protein characterization methods and technologies and more rigorous quality and safety requirements. Protein quantification is an indispensible part of the drug development and production process, from drug discovery through large-scale manufacture and release testing.
However, the development of a precise, robust, and reproducible method remains a key challenge. This is particularly important for manufacturers of protein therapeutics where regulatory standards must be rigorously maintained, and the characterization of both the manufactured products and essential reference standards need to be established with high accuracy.
During biologic drug manufacture, proteins must be tested at all stages to meet GMP standards and all analytical tests must be fully validated. Currently, no single method of protein quantification can determine the true protein concentration for all proteins in every type of buffer—primarily as a result of the large variety/diversity of protein structures and physiochemical properties.
As a result, the most suitable assay or combination of assays must be determined for each product. Assays commonly used for protein quantification include enzymatic activity determination, ion exchange chromatography, quantitative gel electrophoresis, cell-based assays, and ELISA.
Choosing the Correct Assay
The choice of a quantification method should be based on several factors, including:
• the stage in the drug production process at which the assay will be used: During the discovery phase, the drug is only available in low quantities, at low purity, and is often not well characterized. During scaleup and manufacture additional factors such as interaction of formulation ingredients must be considered.
• the goal of the protein quantification approach—whether the drug substance alone or the total protein content of the sample needs to be quantified.
• the individual properties of the protein: It is important to consider parameters such as the solubility, molecular weight, isoelectric point, amino acid composition, stability, and homogeneity of the protein.
Buffer components and other excipients must also be taken into account. For example, low solubility of the protein in aqueous solutions can make the use of detergents or chaotrope reagents necessary, which themselves interfere with many protein quantification assays.
Choosing the best protein quantification assay is not straightforward and requires experience in the field. In most cases multiple assays have to be used, and each of these must be optimized and validated for each specific part of the testing.
Validating an Assay
Validation of all analytical methods is a regulatory requirement. Risk-analysis techniques such as FMEA (failure mode and effect analysis) have proven to be useful in this regard. Validation involves a risk assessment step that is followed by a close analysis of the robustness of each assay. As each protein requires a tailored quantification method this will to some extent determine the required validation process.
To illustrate the complexity of protein quantification, and the validation process, Protagen validated an assay for protein quantification according to ICH guideline Q2(R1), using a tumor lysate as a model matrix and bovine serum albumin (BSA) as a model protein. According to this ICH guideline, several parameters have to be validated for a quantitative assay, outlined in Table 1, with definitions in Table 2.
The aim was to develop a robust assay that is capable of quantifying the total protein content of a sample in a wide variety of buffers. The assay chosen was a modified protein-amidoblack-complex precipitation using triplicate determinations. The modifications included the introduction of a 96-well plate fitted with a membrane bottom, which allowed the reproducible isolation of the protein-dye precipitate.
The quantification was carried out relative to a BSA standard curve. To evaluate critical steps a FMEA was performed.
One of the critical steps identified was the risk of using commercially available BSA without checking the actual concentration of each lot. To overcome this, an NIST standard was employed to check the normal protein concentrations for the calibration curve. Another critical point was identified in the use of the multiwell plates for the assay—each lot of plates needed to be checked for a homogeneous read-out in all well positions.
Having addressed these and other outcomes of the FMEA, the robustness of the assay was considerably increased. Therefore, the validation characteristics of linearity, accuracy, precision (both repeatability and intermediate precision), detection limit, quantification limit, and range could be determined.
Robustness and Specificity
Here, the main concern was to measure only the protein content of the sample and to avoid the influence of buffer components on the quantification results. Proof of specificity was achieved by comparing the results obtained from a placebo (dilution buffer), a spiked placebo, and a sample solution. The buffer composition was shown to have no influence on the quantification results for the buffers tested (Figure). In each case, the concentration of the spiked protein was correctly determined.
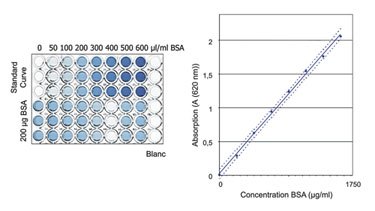
Validation of a protein quantification assay.
Linearity and Accuracy
To determine the linearity, the absorption of eight BSA samples over a range of 100–1,750 µg/mL was measured. The resulting optical density was shown to be directly proportional to the protein concentration in this range.
Accuracy was shown by spiking tumor lysates of known concentration with defined amounts of BSA and subsequently performing a recovery calculation. The acceptance criterion of 100 ±5% recovery was well met.
Precision
The coefficient of variation was shown to be less than 5% for hexaplicate analyses using 500 µg/mL Jurkat cell extractions as sample. This shows the precision of the assay.
Detection limit and quantification limit (QL) were calculated from the standard deviation of a hexaplicate blank (dilution buffer) measurement as well as from the linear dilution curve. The QL was determined to be between 40–100 µg/mL.
Specific Layout
The validation of this assay has enabled the development of a specific plate layout that is now used for all protein quantification analyses. This layout includes the standard curve in triplicate, the samples in triplicate, and negative controls. One major criterion for accepting the results of the assay is the quality of the reference curve, which is measured by the correlation factor and the slope of the linear fit. The data analysis is carried out using a validated Excel spreadsheet.
In summary, the protein quantification outlined here is linear, accurate, and precise, at least within the range of 100–1, 750 µg/mL BSA. This leads to a robust and validated method for protein quantification. The validation shows the main advantages of this quantification method compared to other assays like Bradford or Lowry, including a good tolerance toward differing buffer compositions and a large linear range.
Protein quantification is one of the seemingly easier tasks in protein characterization. It is clear, however, that even a simple assay can be complex when it comes to robustness and validation.
Katja Aschermann, Ph.D. ([email protected]), is head of quality management, Martin Blüggel is COO, and Andreas Wattenberg, Ph.D., is director of analytics at Protagen. Web: www.protagen.de.

