Single-cell RNA sequencing (scRNA-seq) emerged to characterize gene expression differences between individual cells derived from a complex tissue, allowing a higher resolution look at mRNA expression than bulk RNA-seq. With each passing week, the lens of single-cell information is delivering new insights into development, cancer, and neurobiology. These techniques are also offering new avenues to approach genetic screens and synthetic biology in a powerful, scalable manner.
To date, the most common scRNA-seq methodologies have been coupled to short-read sequencing platforms that offer quantification of gene counts either based on the 3′ end tags or sparse information across the entire length of the transcripts, neither of which can reveal contiguous mRNA sequences and the associated isoform-specific open reading frames which often encode proteins with different functional properties.
The PacBio RNA sequencing solution, called the Iso-Seq method,1 uses single-molecule, real-time (SMRT) sequencing technology to produce full-length, highly accurate long reads that can be used in combination with short-read RNA-seq approaches. The Iso-Seq reads can span the entire 5′ to 3′ end of a transcript, allowing a high-resolution view of isoform biology without the need for assembly. Although the Iso-Seq method has been used for hundreds of studies to date and has become a gold standard for genome annotation, researchers have only recently taken advantage of the long reads for single-cell isoform analysis.
Many single-cell platforms use a similar molecular biology workflow to the Iso-Seq method, with reverse transcription copying the mRNA from the polyA tail at the 3′ end to a template switch reaction to tag the 5′ end. The unique aspect of a single-cell sequencing library is that it also contains cell barcode and unique molecular index (UMI) information to confidently assign reads to individual cells. The most common plate-based and microfluidic single-cell systems encode this additional information into the reverse transcription primer.
Last year, scientists at Weill Cornell Medicine showed that one of these systems can be adapted to sequence full-length isoforms in the mouse brain.2 More recently, in collaboration with scientists at the University of California, San Francisco, we demonstrated that the Dolomite Nadia platform can perform Drop-Seq aimed at studying isoforms expressed in cells from human and chimpanzee cerebral organoids.3
Single-cell library preparation and sequencing
Although PacBio does not have a specific single-cell partner or system recommendation, in principle, practically any single-cell platform should be compatible with single-cell Iso-Seq library preparation so long as that platform generates cDNA. For the Iso-Seq method to achieve full-length cDNAs, it is recommended to use a template-switching oligo. This is a common technique and is currently used in single-cell platforms and PacBio’s current bulk Iso-Seq methods.
Generating a single-cell Iso-Seq library is then a two-step process. First, at the point in the protocol where the cDNA is usually sheared for short-read workflows, the intact RT-PCR product is instead amplified further to yield more cDNA material for PacBio template preparation and sequencing. This amplification is achieved using PCR primers specific to a single-cell platform, and these sequences should be obtained from the platform provider. Second, this amplified cDNA is converted to a sequencing library using the PacBio SMRTbell Express Template Prep Kit 2.0.
For best analytical results, it is recommended to generate short- and long-read data on the same single-cell library. The additional amplification can easily yield sufficient material to generate sequencing libraries for both Illumina and PacBio runs. On the PacBio Sequel II System, a target of >160 ng DNA is recommended. A sizing platform such as the Agilent Bioanalyzer is a useful tool for checking the quality of the amplified cDNA; the mean size of a high-quality single-cell cDNA library is usually ~1.5 kb with molecules that stretch into the 5–6-kb range.
Single-cell Iso-Seq bioinformatics analysis
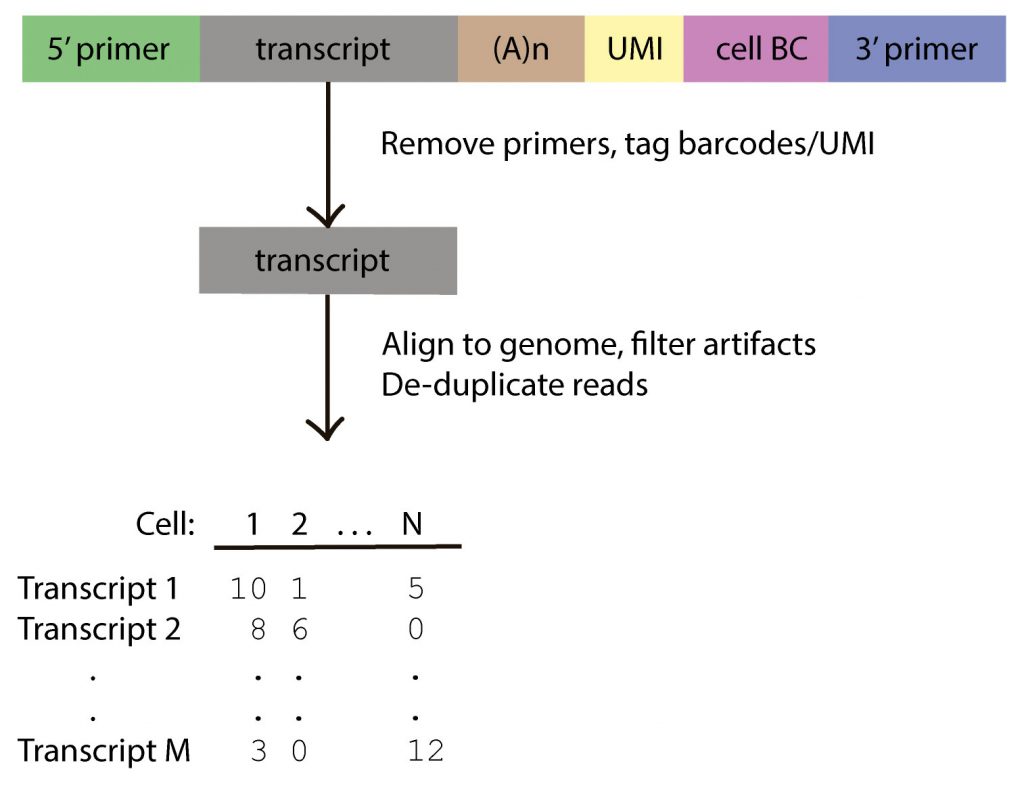
Bioinformatics analysis for the single-cell Iso-Seq method follows a similar procedure to short-read single-cell data (Figure 1). The basis of a single-cell Iso-Seq read is a highly accurate long read—that is, a high-fidelity (HiFi) read—using the circular consensus sequencing (CCS) mode that generates Q20 (>99%) accurate reads,4 making cDNA primer removal and UMI/cell barcode detection a reliable process:
- Generate HiFi reads using the CCS sequencing mode
- Remove 5′ and 3′ cDNA primers
- Clip UMI and cell barcodes
- Remove polyA tails—after this step, the read consists only of full-length reads with the proper strand polarity Align full-length reads to the genome and classify against reference annotation
- De-duplicate reads based on UMI, barcode, and gene information
- Generate gene count matrix or isoform count matrix, which can then be used with popular single-cell pipelines such as Seurat and Scanpy
It is recommended that matching short-read data be generated, which can be independently run through standard single-cell pipelines to identify cell-type information. In this way, even at lower throughput, the Iso-Seq data will complement the short-read data by matching back to the same cell barcodes.
Detecting novel isoforms with full-length single-cell sequencing
In the poster we presented at Advances in Genome Biology and Technology,3 we showed the use of the SQANTI2 software to classify full-length, single-cell data against the GENCODE annotation (Figure 2). Not only were a significant portion of the full-length transcripts novel, but the majority of them were predicted to be coding.
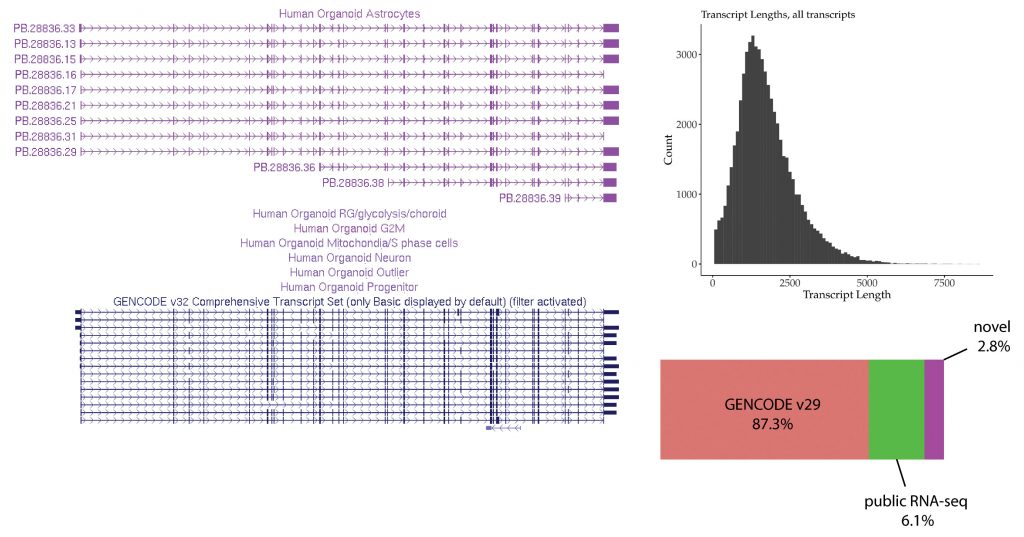
The SQANTI2 software additionally used orthogonal evidence to validate that the transcripts were full length: more than half of the known and novel isoforms have the 5′ end fall within 50 bp of a CAGE peak, and most of the 3′ ends have a canonical polyadenylation motif signal. The Intropolis database—a massive RNA-seq junction database collected from more than 21,000 public RNA-seq data sets—helped to validate novel junctions identified in the full-length Iso-Seq data. Finally, SQANTI2 filtered out cDNA artifacts that were not well supported by orthogonal data, producing the final set of high-confidence full-length transcripts that revealed complex alternative splicing events in a cell-type-specific manner.
Conclusion
Combining single-cell technology with the ability to sequence full-length transcripts using highly accurate long reads has the potential to reveal cell-to-cell heterogeneity at the isoform level. The field of single-cell sequencing is moving fast, and as molecular methods continue to improve and evolve, long accurate reads generated by SMRT sequencing will be of increasing importance as it complements existing short-read approaches.
Single-cell research is already making progress in the human disease space, and long reads offer unique advantages: a recent study by scientists based at the Fred Hutchinson Cancer Research Center showed how single-cell infection combined with targeted long reads revealed the astounding mutational diversity that occurs in the influenza virus.5 This study and those to come will provide a higher resolution look at biology and disease through the precision lens of single-cell, single-molecule sequencing.
References
1. Pacific Biosciences website. Applications: RNA Sequencing. Accessed
December 4, 2019.
2. Gupta I, Collier PG, Haase B, et al. Single-cell isoform RNA sequencing
characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018; 36: 1197–1202. doi:10.1038/nbt.4259.
3. Tseng E, Underwood JG, Bhuduri A, et al. Single cell isoform sequencing
(scIso-Seq) identifies novel full-length mRNAs and cell type-specific expression. Poster presented at: Advances in Genome Biology and Technology;
February 22–March 2, 2019; Marco Island, FL.
4. Pacific Biosciences website. SMRT Science: SMRT Sequencing Modes.
Accessed December 4, 2019.
5. Russell AB, Elshina E, Kowalsky JR, et al. Single-Cell Virus Sequencing of
Influenza Infections That Trigger Innate Immunity. J. Virol. 2019; 93(14): e00500-19. doi:10.1128/JVI.00500-19.
Elizabeth Tseng, PhD, and Jason G. Underwood, PhD, are principal scientists at Pacific Biosciences.
