G protein–coupled receptors (GPCRs) play crucial roles in the regulation of a wide variety of physiological processes and represent one-third of targets with clinically prescribed drugs. Yet, it remains the case that currently approved drugs act at only ~16% of the GPCRome.1 Expanding the therapeutic GPCR target space will require the development of innovative screening approaches that incorporate novel concepts in GPCR biology and pharmacology.
Classically, GPCR-mediated signal transduction was thought to occur via linear signaling pathways, with a given GPCR selectively engaging a single heterotrimeric G protein family defined by the sequence and function of the Gα subunit. The human genome codes for 16 Gα proteins that are divided into four major families (Gαs, Gαi/o, Gαq/11, and Gα12/13). Following activation, these proteins each engage different downstream effectors that ultimately provoke different signaling (and thus biological) outcomes.
Two key complementary discoveries in the last decade, namely pluridimensional efficacy and biased agonism,2,3 have revolutionized our understanding of GPCR biology and the pharmacology of ligands targeting these receptors. Contrary to the classical view of GPCR signaling, it is now evident that these receptors can exist in multiple active (that is, “on”) states and engage multiple (potentially cross-talking) downstream signaling cascades by directly coupling to both heterotrimeric G proteins and non-G protein effectors (β-arrestins). Further, a given drug acting on a receptor can display different (sometimes reversed) efficacies toward the various pathways coupled to the receptor. Drug efficacy is thus considered to be a pluridimensional parameter.
An extension of this observation is described by the concept of biased agonism (or functional selectivity), which posits that structurally dissimilar ligands can stabilize unique receptor conformations with each conformation differentially coupling to downstream effectors. Accordingly, different ligands of a given receptor may impart distinct signaling attributes (or “signatures”) and biological properties to the receptor.
The promise of biased agonism lies in its applicability for the development of GPCR ligands that selectively engage pathways with therapeutic benefit while inhibiting or remaining inactive toward those producing deleterious outcomes. Ultimately, biased ligands may display superior therapeutic profiles in many disease areas and indications.
To fully explore the potential of functional selectivity in drug discovery and development, it becomes essential to have high-throughput screening (HTS) technologies enabling an exhaustive sampling of the various effectors that can be engaged by a given receptor, providing receptor- and ultimately ligand-specific signaling signatures. Yet, current high-throughput methodologies used for GPCR and ligand signaling profiling largely involve the measurement of second messenger production (that is, inositol phosphate (IP), Ca2+, cAMP). Many of these receptor-distal readouts rely on biological responses that can be modulated by multiple (often cross-talking) downstream signaling pathways. Consequently, second messenger levels alone are not directly indicative of a ligand’s activity or efficacy on a specific heterotrimeric G protein pathway, and their use for ligand profiling may thus lead to an incomplete view about a ligand’s signaling signature.
Overview of the bioSensAll™ GPCR platform
bioSensAll™ is a homogenous, live-cell, enhanced bystander bioluminescence resonance energy transfer (ebBRET)-based biosensor platform that enables the real-time monitoring and mapping of the signal transduction pathways engaged upon GPCR activation. In contrast to commonly used second messenger and reporter assays, bioSensAll™ technology revolves around the measurement of receptor-proximal events directly linked to GPCR activity: heterotrimeric G protein activation and β-arrestin coupling.
Unique to this approach to ligand profiling is the ability to directly quantify receptor coupling to specific heterotrimeric G protein subtypes and β-arrestin isoforms without the need to tag the receptor or G proteins (except for Gαs). To date, this HTS-compatible platform includes biosensors for the activation of 14 distinct heterotrimeric G proteins (Gαs(L), Gαs(S), Gαq, Gαi1, Gαi2, Gαi3, GαoA, GαoB, Gαz, Gα11, Gα12, Gα13, Gα14, and Gα15/16) as well as sensors for the engagement of β-arrestins 1 and 2.
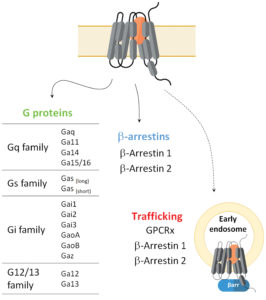
These sensors can also be used to analyze spatial aspects of GPCR signaling by enabling the study of receptor and/or effector translocation and activity at endosomal compartments (Figure 1). These sensors thus offer a significant level of data granularity on receptor and ligand signaling repertoires.
Altogether, the GPCR downstream signaling signatures obtained via this biosensor platform may provide insight into the therapeutic efficacy and safety of pharmacologically active compounds. Accordingly, characterizing the signaling profiles of GPCR ligands may help better inform pharmaceutical firms in their lead selection process and ultimately reduce attrition risks.
Functional details of the bioSensAll™ assay
Unlike conventional BRET assays, ebBRET exploits the ability of luciferase and GFP from Renilla reniformis to self-associate with mild affinity and to optimally transfer energy, resulting in improved assay windows and sensitivity.4 Consequently, these naturally interacting chromophores were exploited to develop new, highly dynamic BRET-trafficking sensors designed to monitor the plasma membrane recruitment of proteins that interact with active (GTP-bound) Gα subunits in a G protein family–selective manner.
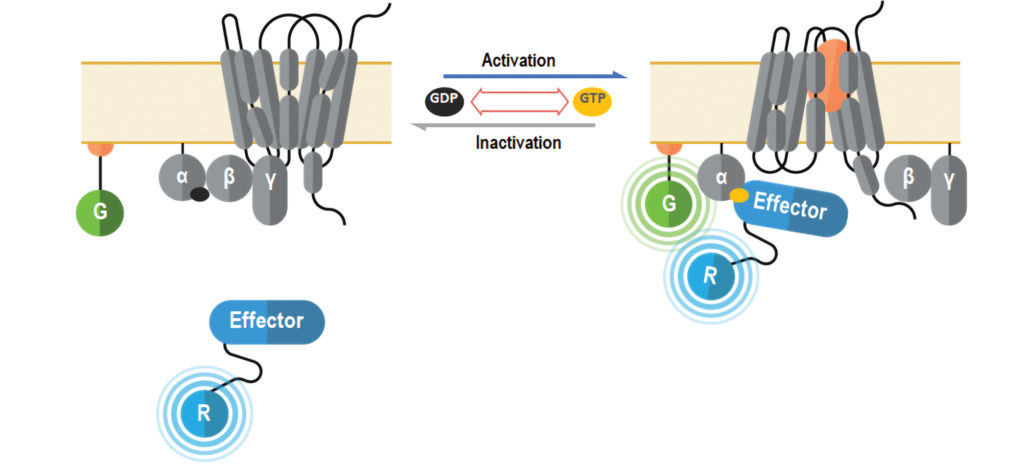
The G protein biosensors constitute a GoLogo motif domain of Rap1GAP (interacts with all Gαi/o/z subunits), a regulator of G protein signaling (RGS) domain of p63RhoGEF (interacts with Gαq/11/14/15 subunits) or an RGS-like domain of PDZ-RhoGEF (interacts with Gα12/13 subunits) fused to Renilla luciferase (RlucII, R in Figure 2). The translocation and recruitment of biosensors to the plasma membrane upon GPCR activation translates into an increased BRET efficiency with a plasma membrane–anchored Renilla green fluorescent protein (rGFP, G in Figure 2). The same plasma membrane translocation principle is used to measure receptor-mediated β-arrestin engagement with RlucII fused directly to either β-arrestin 1 or β-arrestin 2.
Case study: Profiling of PTH/PTH1R signaling
Parathyroid hormone (PTH)/parathyroid hormone–related protein receptor (PTH1R) is a family B GPCR involved in the regulation of skeletal development, bone turnover, and calcium homeostasis. Indeed, PTH(1–34) (teriparatide) is used clinically to stimulate restoration of bone mass for the treatment of osteoporosis. However, the clinical applicability of PTH-based osteoporosis therapies is limited by their poor pharmacokinetics (require daily injection) and their propensity to produce hypercalcemia. Such hypercalcemia is related to the tight coupling of anabolic and catabolic effects of PTH in bone in addition to its capacity to stimulate the rates of renal calcium reabsorption and 1,25-dihydroxyvitamin D3 production.5
Activation of PTH1R by PTH has been shown to promote receptor association to both G proteins and β-arrestins. Interestingly, functionally selective (that is, pathway-biased) PTH1R ligands have displayed the potential to uncouple the beneficial anabolic effects from catabolic effects of PTH1R activation in bone. Modification of PTH N- or C-termini (via truncations or introduction of limited amino acid changes) has been demonstrated to alter receptor signaling and may guide the development of functionally selective PTH1R ligands.6 Such ligands may afford advantages in displaying reduced catabolic and calciotropic side effects and improved pharmacokinetics.
Herein, we demonstrate how bioSensAll™ biosensors were used to characterize and differentiate the signaling signatures of two PTH1R ligands: PTH(1–34) (N-terminal 34 amino acids of PTH shown to possess all of the known properties of the native PTH) and the PTH analogue Tyr1-PTH. Both ligands were profiled on various G protein and β-arrestin ebBRET biosensors (Figure 3).
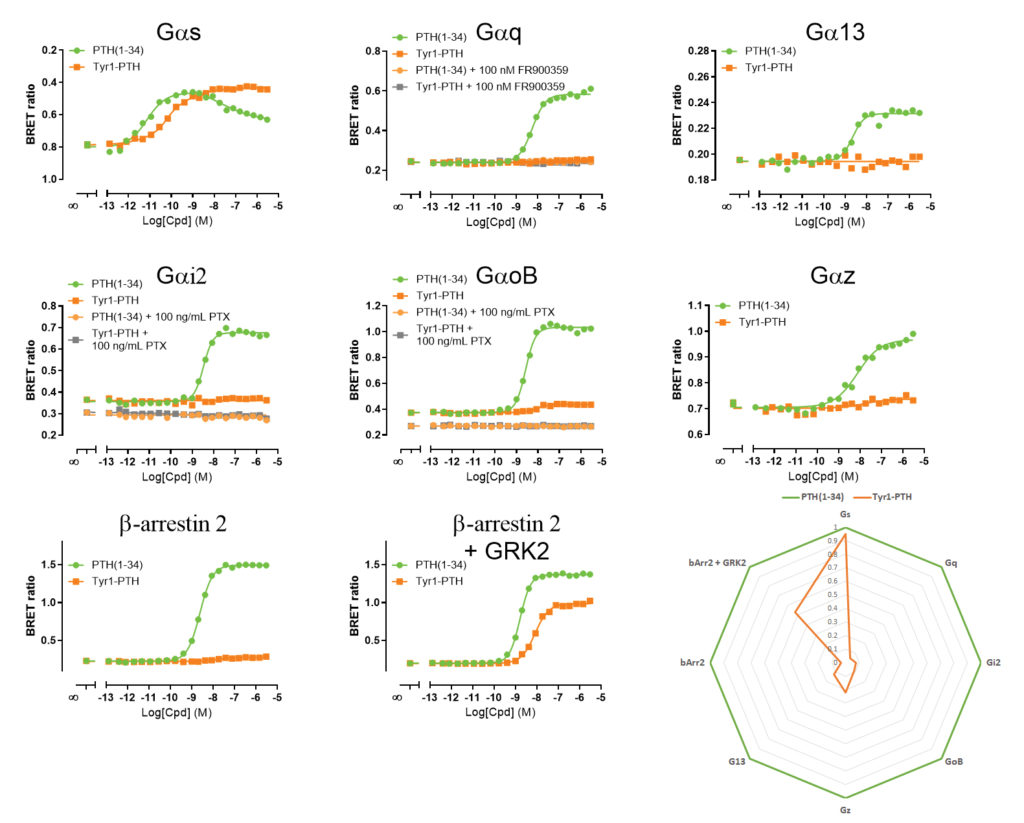
Our investigation revealed that in response to its endogenous
ligand, hPTH1R engaged Gαs, Gαq, Gαi-family G proteins (Gαi2, GαoB, Gαz), Gα13, and β-arrestin 2. Further, pretreatment with pertussis toxin completely blocked Gαi/o activation while pretreatment with the Gαq/11-selective inhibitor FR900359 ablated Gαq engagement.
Interestingly, Tyr1-PTH activated Gαs (full agonist) only while remaining relatively ineffective at stimulating the other pathways engaged by PTH(1–34). Interestingly, co-expression of GPCR kinase 2 (GRK2), a key regulator of GPCR signaling in bone, dramatically promoted Tyr1-PTH’s efficacy to engage β-arrestin 2 while not affecting PTH(1-34) activity on this pathway.
Conclusion
The bioSensAll™ biosensor platform provides a qualitative and quantitative assessment of a receptor’s complete signaling repertoire and allows for the identification and characterization of functionally selective ligands in a rapid, simple, and homogenous assay. Of note, all assays are HTS-compatible and performed using native, untagged receptor. Further, all biosensors (G protein and β-arrestin) are based on a common assay principle and not a combination of distinct assay formats that may differ in kinetics and/or signal amplification. The platform allows for a more direct comparison between signaling outputs, minimizing artefacts in the detection of biased signaling and functionally selective ligands. In addition, the platform allows for the integration of accessory proteins (such as GRK2) into the assay workflow to evaluate their effects on receptor signaling and ligand functional selectivity. Such information may reveal relevant biology and pharmacology critical to translating in vitro ligand functional selectivity to therapeutic efficacy in vivo.
Arturo Mancini, PhD, is scientist and Laurent Sabbagh, PhD, is associate director at Domain Therapeutics North America.
References
1. Hauser AS, Gloriam DE, Brauner-Osborne H, Foster SR. Novel approaches leading towards peptide GPCR de-orphanisation. Br. J. Pharmacol. 2020; 177(5): 961–968.
2. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012; 52: 179-97.
3. Costa-Neto CM, Parreiras-E-Silva LT, Bouvier M. A Pluridimensional View of Biased Agonism. Mol. Pharmacol. 2016; 90(5): 587–595.
4. Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA. Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 2016; 7: 12178.
5. Luttrell LM, Maudsley S, Gesty-Palmer D. Translating in vitro ligand bias into in vivo efficacy. Cell. Signal. 2018; 41: 46–55.
6. Cupp ME, Nayak SK, Adem AS, Thomsen WJ. Parathyroid hormone (PTH) and PTH-related peptide domains contributing to activation of different PTH receptor-mediated signaling pathways. J. Pharmacol. Exp. Ther. 2013; 345(3): 404–418.
