
With an adeno-associated virus (AAV)-based product now licensed by the U.S. Food and Drug Administration for treatment of spinal muscular atrophy in newborns, the race is on to fully exploit AAV-based gene therapy, a race that is beset by hurdles. One of the hurdles, downstream processing, is often higher than developers expect because they must deal with two AAV product–specifific challenges. First, developers must cope with stray chromatin, the chromosomal debris in lysates left by dead host cells. Second, developers must separate empty capsids from the full capsids containing the gene therapy load.
Chromatin elimination and extraction
The presence of chromatin is uniquely a problem of in vitro cell culture production. Dead cells in vivo are removed by phagocytes and transported elsewhere for lysis and recycling, but cell culture lacks phagocytes. Chromatin degradation in cell culture begins during apoptosis and continues through secondary necrosis but does not go to completion. The contents of dead cells accumulate over the life of the culture. Nucleosomal arrays, nuclease-resistant subpopulations of chromatin, develop stable associations with RNA and nonchromatin host proteins that persist for weeks to months after harvest.1 The chromatin burden is compounded if living cells are lysed.
Chromatin is not simply a contaminant to be removed. It represents a heterogeneous class of contaminants that substantially impairs the function of all purification tools. Detailed process-relevant studies to date have mostly addressed monoclonal antibodies produced by mammalian cells,1–4 but chromatin is produced by all eukaryotic cells. Its downstream liabilities are largely the same for all biologics produced by cell culture, except that the magnitude of those liabilities is proportional to the ratio of chromatin to product in any given feed stream.
Although chromatin constitutes less than 10% of the contaminant mass, it causes more than 90% of the problems in purification. Its DNA component is extremely electronegative, and its histone protein component is highly electropositive. Both components further participate in hydrophobic, hydrogen bonding, metal affinity, and van der Waals interactions. These properties make chromatin sticky from a chemical perspective and cause it to depress the effectiveness of all purification tools and methods—chromatography, filtration, and precipitation.
The strong multimodal associations between DNA and histone proteins meanwhile protect the DNA component against lysis by nuclease enzymes, so complete lysis is unachievable. Beyond burdening purification, this inability of nucleases to fully digest host DNA causes analytical methods to underestimate DNA content. Typically, 5–20% of DNA remains after nuclease treatment, still strongly associated with histone proteins, in two subclasses. The first includes partially intact nucleosomal arrays ranging from about 10 nm to more than 400 nm. The second includes histone–DNA fragment associations ranging from 2 nm to 10 nm. Both form metastable secondary associations with RNA, both with nonchromatin host proteins and with each other.
On the positive side, chromatin elimination in advance enables purification performance to rebound to the levels process developers expect. Chromatography binding capacity increases 20–70%, depending on the method. Host protein removal becomes 100 times more effective, host DNA removal becomes more than 1000 times more effective, and product recovery increases 10–50%.3,4 Reproducibility also improves. Uncontrolled lot-to-lot variation in chromatin content among harvests and lysates, due in part to chromatin’s effects on purification efficiency, translates to excess process variation. Advance removal eliminates that variation.
Many methods for chromatin reduction from antibody cell culture harvests have been described, including solid-phase adsorbents that preferentially bind histones or DNA and a variety of flocculating agents, but the overall choice of conditions needs to be customized to each individual product class. For example, many of the agents and conditions applied to IgG cause major product losses with IgM, while IgM-optimized methods support less effective chromatin removal than IgG-optimized methods.
Materials and conditions for advance chromatin removal from AAV lysates can be expected to be distinct from methods for antibodies, perhaps dramatically so, since the methods used for IgG harvests also remove up to 9 logs of virus, including small non-lipid-enveloped species.3,4 As AAV-directed methods begin to emerge, industry experience will reveal the magnitude of adjustment that may be required to accommodate the respective surface chemistries among different AAV serotypes.
Figure 1 shows results from an experimental chromatin extraction process applied to a lysate containing AAV8 grown in SF9 cells. Extraction was performed by adding CIMapshere™ AAV particles, which have a high affinity for DNA. After incubation, the particles were removed by filtration. The assay employs size exclusion chromatography (SEC) with simultaneous monitoring by UV at 260 nm and 280 nm. Ratios of 260 absorbance to 280 absorbance greater than 1.0 tend to correlate with high DNA content. Note the broad size distribution of chromatin remnants in the nuclease-treated sample at left. Simultaneous monitoring of tryptophan fluorescence elevates sensitivity of protein detection and enables visualization of the AAV population, even against the backdrop of UV-absorbing host cell contaminants. Most of the DNA-associated contaminants are removed by the extraction.
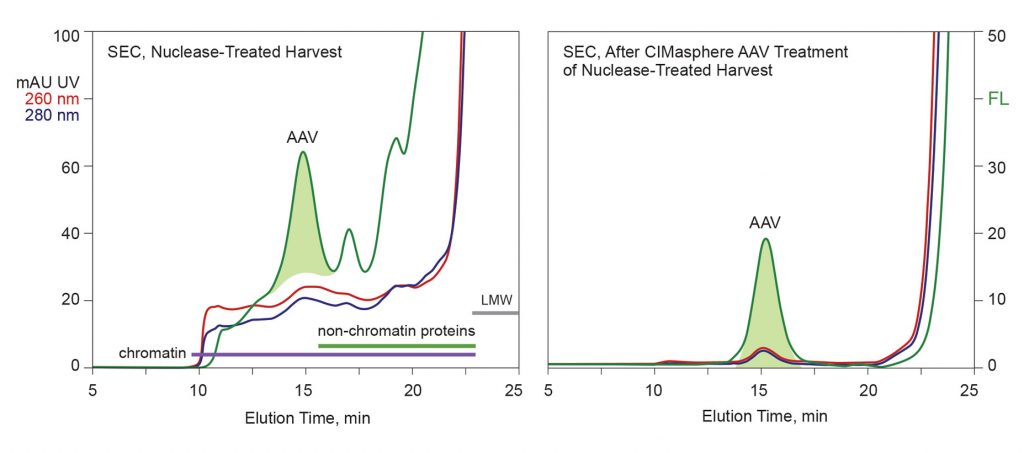
Figure 2 highlights one of the key benefits of advance chromatin extraction. Suspending interference with purification tools enables economical capture options, like cation-exchange chromatography, to offer good capacity and a high purification factor. Persistent chromatin contamination indicates that extraction does not reach completion. The chromatogram shows 260-dominant DNA-containing contaminants eluting before the AAV. Polyacrylamide gel electrophoresis reveals their primary protein components as histones H1 and H3. The AAV population nevertheless emerges highly purified. Cation-exchange chromatography is broadly applicable to AAV. All serotypes can be purified with modest adjustments to platform buffers and elution conditions.
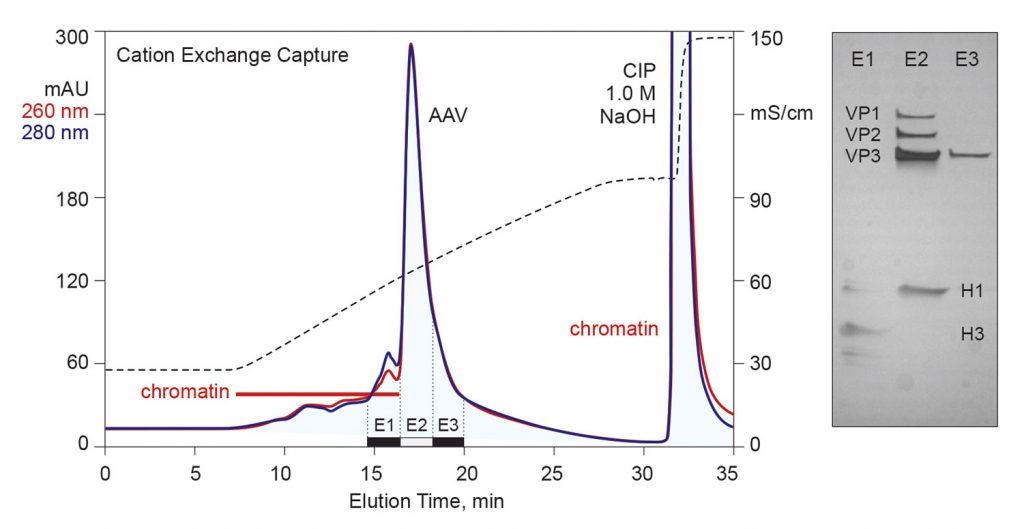
Monolithic chromatography media are especially well suited for AAV purification for both technical and practical reasons. Mass transport through monoliths is convective. One important benefit of this is that they easily support flow rates that are more than 10 times faster than those supported by columns packed with porous particles.
Higher flow rates translate to shorter process time and higher manufacturing productivity. In many cases, this makes it unnecessary to conduct a preparative tangential flow filtration step to concentrate a sample before loading the capture column.
A corollary of convective mass transport is that both capacity and separation performance are independent of flow rate. This is very important across scale-up increments because it means that the linear flow rate need not be held constant. Scale-up becomes a simple matter of maintaining the volumetric ratios of sample, buffer, and column.
Separation of empty and full capsids
At present, separation of empty AAV capsids from full capsids often employs ultracentrifugation, which consistently yields 95% elimination of empties. However, scalability is difficult, the equipment is expensive, and the technique is intolerant of even minor operator errors. These issues continue to push the industry toward the more familiar approach of chromatography-based fractionation, but this represents a very significant challenge. The degree of surface differentiation between empty and full capsids is modest at best and varies among serotypes.
Figure 3 compares the capacity of two chromatography methods for fractionation of empty and full capsids. The left-hand panel illustrates a chromatogram from a salt gradient separation on a strong anion-exchange monolith. Empty capsids can be distinguished from full capsids by the ratio of UV absorbance at 260 nm and 280 nm. Proteins dominantly absorb UV at 280 nm; nucleic acids, at 260 nm. Full capsids generally exhibit a 260/280 ratio modestly greater than 1.3. The fraction corresponding to the black bar was estimated by fluorescent analysis to contain 95% full capsids. Published comparisons of results between full capsids prepared by ultracentrifugation and anion-exchange chromatography show good agreement.5
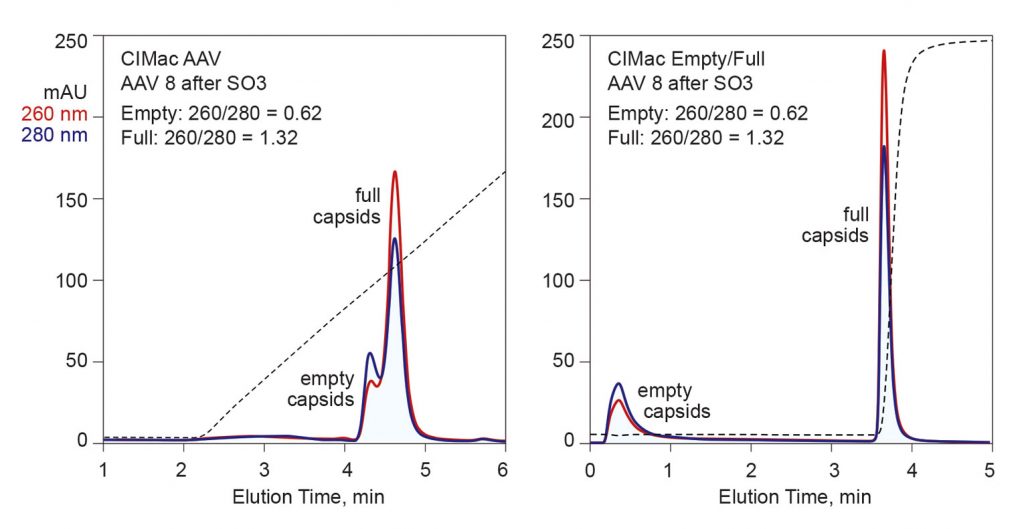
Anion exchange represents a good start, but there is room for improvement. The overlap between empty and full peaks means that some full capsids need to be sacrificed to achieve the best elimination of empties using this method. Another issue is that the separation requires exposure to very high pH values (pH 9 and above), where capsid stability begins to become a potential concern. Scale-up to industrial chromatography skids with linear-gradient capability is simple and robust, but performance may be compromised on chromatography skids with only step-gradient capability.
The right-hand panel of Figure 3 illustrates results from a multimodal monolith developed expressly to exploit the small differences in the surface chemistry of empty and full capsids, and particularly to suspend the limitations of strong anion exchangers. Empty capsids flow through the column under sample application conditions, then full capsids are eluted in a step to near-physiological conditions. Preliminary indications are that higher capacity can be obtained by binding both empties and fulls, eluting empty capsids in one step, then full capsids in another. The separation can undergo scale-up on linear- or step-gradient skids without compromising either performance or reproducibility.
Pete Gagnon is the chief scientific officer of BIA Separations.
References
1. Tan L, Yeo V, Yang Y, Gagnon P. Characterization of DNA in cell culture supernatant by fluorescence-detection size-exclusion chromatography. Anal. Bioanal. Chem. 2015; 47: 4173–4181.
2. Mechetner L, Sood R, Nguyen V, Gagnon P, Parseghian MH. The effects of hitchhiker antigens co-eluting with affinity-purified research antibodies. J. Chromatogr. B 2011: 879: 2583–2594.
3. Gagnon P, Nian R, Lee J, Tan L, Abdul Latiff SM, Lim CW, Chuah C, Bi X, Yang YS, Zang W, Gan HT. Nonspecific interactions of chromatin with immunoglobulin G and protein A, and their impact on purification performance. J. Chromatogr. A 2014; 1340: 68–78.
4. Nian R, Zhang W, Tan L, Lee J, Bi X, Yang Y, Gan HT, Gagnon P. Advance chromatin extraction improves capture performance of protein A affinity chromatography. J. Chromatogr. A 2016; 1431: 1–7.
5. Fu X, Chen WC, Argento C, Clarner P, Bhatt V, Dickerson R, Bou-Assaf G, Bakhshayeshi M, Lu X, Bergelson S, Pieracci J. Analytical Strategies for Quantification of Adeno-Associated Virus Empty Capsids to Support Process Development. Hum. Gene Ther. Methods 2019; 30: 144–152.
