
February 15, 2016 (Vol. 36, No. 4)
Revolutionizing Whole-Genome Screening, Advancing Target Identification
Thanks to next-generation sequencing (NGS), we are starting to understand the mutational changes that occur across the board in the cancer genome. With this knowledge comes potential—novel mutated genes and the proteins that they encode are candidates for prognostic markers and/or new drug targets. However, taking these initial findings all the way through to the clinic is an enduring challenge.
RNAi technologies initially looked as if they might provide a new avenue for more effective drug discovery, but off-target effects and variable knockdown efficiencies make this a fraught approach unless stringently controlled. The advent of gene editing using CRISPR-Cas9—essentially repurposing a primitive adaptive immune response in bacteria—provides a new and powerful tool to interrogate gene function on a genome-wide level. From our current vantage point, the contribution of this technology to drug discovery looks like it will be substantial, primarily because the off-target effects appear to be fewer in CRISPR-Cas9 target ID screens.
Introducing Indels
CRISPR-Cas9 screens make use of two aspects of the CRISPR machinery: short guide RNAs (sgRNAs) and the Cas9 nuclease. Cas9 binds to the sgRNA, which in turn can bind with the matching genomic DNA sequence upstream of a protospacer adjacent motif (PAM). PAMs ensure that Cas9 will bind to the RNA:DNA hybrid and introduce DNA double-strand breaks. These are repaired by nonhomologous end joining (NHEJ), which frequently results in small nucleotide insertions and/or deletions (indels) that can lead to the interruption of the normal reading frame, thereby disrupting gene function. sgRNAs libraries combined with Cas9 expression can be used to generate gene knock-outs with high on-target specificity.
Taking a Pooled Approach
Using lentiviral transduction to deliver both the Cas9 nuclease and sgRNAs into cells, we have adapted a pooled-based screening protocol to enable us to examine the effect on cell survival of knocking out thousands of individual genes. The sgRNAs are designed to direct Cas9 to exons at the start of the open reading frame such that any out-of-frame indels will result in gene disruption.
Screens are carried out at a suitable coverage level (approximately 300 cells per sgRNA or higher) to maintain representation of every sgRNA within the library, and cells are infected with lentivirus at low multiplicity of infection to ensure single integration events. Antibiotic selection is used to eliminate non-transduced cells prior to the start of the screen and a baseline cell pellet is harvested to allow library representation to be assessed ahead of the screening phase. This initial time point also acts as a comparator for the pellet collected at the end of the screen, such that sgRNA loss and gain over the time of the screen can be established.
After antibiotic selection, cells are seeded into different treatment conditions and maintained for at least 12 population doublings to allow gene editing to progress to completion and the resulting phenotypic effects to be manifest. Genomic DNA is extracted from the cell pellets and guide RNA frequency resolved by NGS. We use a dedicated CRISPR–Cas9 screen analysis platform, adapted from the MAGeCK workflow (Li et al., 2014), which enables individual sgRNAs and gene hits to be ranked (Figure 1).

Figure 1. CRISPR-Cas9 screen workflow
Resistance Is Discoverable Ahead of the Clinic
Approximately 50% of melanomas harbor an activating mutation in the serine/threonine kinase BRAF, and the drug vemurafenib has shown impressive response rates in people that carry the BRAFV600E activating mutation. However, patients with this mutation eventually relapse owing to drug resistance. Many of the mechanisms that underlie this resistance have been documented and, as a proof of concept, we repeated a published genome-wide positive selection screen to identify vemurafenib resistance factors. The GeCKOv2 genome-wide library, which contains 6 guide RNAs against 19,050 genes (Sanjana et al., 2014), was transduced into BRAFV600E mutant A375 melanoma cells and the cells were treated with vemurafenib. This drug had a cytostatic effect on cells, allowing the subset containing resistance-conferring gene-editing events to accumulate.
A comparison of the abundance of each guide RNA at the start and end of the screen was used to assess whether drop out or enrichment of guide RNAs had occurred over the course of the screen. Of those, 228 guides were shown to be >100-fold enriched, with several sgRNAs targeting the same genes. Using our analysis platform, the highest-ranking targeted genes (MED12, NF1, CUL3, NF2, TADA2B, and TADA1) were genes whose loss is known to confer resistance to vemurafenib (Figure 2A).These findings recapitulated previously published data (Shalem et al., 2013; Huang et al., 2012; Whittaker et al., 2013).
Interestingly, when sgRNAs for each of these hits were evaluated individually, it was clear that although the majority of guides were enriched with vemurafenib treatment, not all guides performed equally well (Figure 2B). This emphasizes that for successful screening the library composition and complexity are key considerations.
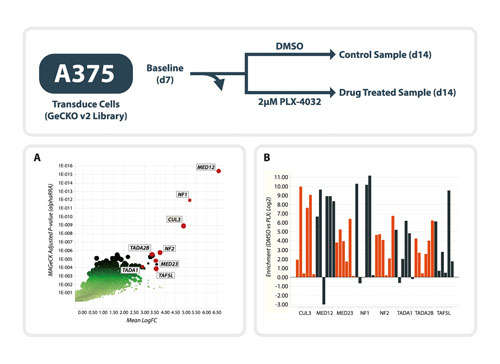
Figure 2. Vemurafenib resistance screen in A375 cells using the whole-genome GeCKOv2 library.
(A) Prosecco plot of screen data. On the y axis, genes are ranked by robust ranking aggregation values for their enrichment after vemurafenib treatment. The mean log2-fold change in sgRNAs targeting the same gene are plotted on the x axis. (B) Efficiency of individual gRNAs against the eight highest ranking genes. For each gRNA, the log2-fold change in sgRNA frequency between vemurafenib and DMSO treatment is plotted.
Increasing the Odds by Halving the Genome
A purine antimetabolite, 6 thioguanine (6-TG), is used in the treatment of leukemia. During DNA replication, 6-TG is incorporated into DNA instead of dGTP, and a small amount is then methylated to generate 6-meTG. During the next replication cycle 6-meTG can base pair with either cystosine or thymine, and both of these mismatches are recognized by the DNA mismatch repair (MMR) system, resulting in a G2M arrest and cell death. The biology of the MMR system and factors that mediate resistance to 6-TG have been extensively studied and, as such, provide an excellent paradigm to test the power of CRISPR-Cas9 resistance screens in haploid cells.
Engineered from HAP1 fibroblasts, eHAP cells are a fully haploid cell line and are particularly suited to CRISPR-Cas9 screens because there is only one copy of any given gene to edit. For this screen, eHAP cells were infected with the whole genome GeCKOv2 library, and following antibiotic selection cells were seeded into either 500 nM 6-TG or vehicle control and maintained in culture to allow sgRNA enrichment and depletion to occur. Based on relative guide abundance, NGS and analysis revealed several genes whose loss of expression conferred resistance to 6-TG treatment (Figure 3A). These included MLH1, MSH2 and MSH6, three genes that encode MMR proteins (Branch et al., 1993; de Wind et al., 1995; Abuin et al., 2000; Buermeyer et al., 1999). Loss of these genes probably prevents mismatch detection, and the cell survives and progresses through the cell cycle, albeit with a higher mutation burden.
To validate these targets as 6-TG resistance factors, MLH1 and MSH6 knockout HAP1 cells were generated using a CRISPR-Cas9 approach. These cells were able to proliferate in the presence of 6-TG, unlike the parental cell line (Figure 3B).

Figure 3. 6-TG resistance screen in eHAP cells using the whole-genome GeCKOv2 library.
(A) Prosecco plot of screen data. On the y axis, genes are ranked by robust ranking aggregation values for their enrichment after 6-TG treatment. The mean log2-fold change in sgRNAs targeting the same gene are plotted on the x axis. (B) Hit validation using engineered knockout cell lines shows that loss of MLH1 and MSH6 confers resistance to 6-TG treatment.
Onwards and Upwards
The application of CRISPR-Cas9 technology to whole-genome screening is revolutionizing our ability to perform target identification experiments and to understand complex biological processes, such as drug resistance. Devoid of the caveats associated with siRNA and shRNA reagents, the hope is that novel targets can be uncovered and rigorously validated using CRISPR-Cas9, and that a pipeline of innovative and validated targets will enter drug discovery programs.
Ceri Wiggins, Ph.D., is team leader and Steffen Lawo, Ph.D., is senior scientist at Horizon Discovery. For more information, write to [email protected].

