
December 1, 2011 (Vol. 31, No. 21)
Combining Specificity and Sensitivity with High Performance for a Multitude of Applications
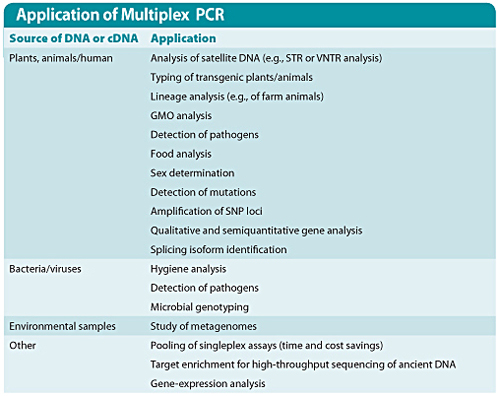
Multiplex PCR is a difficult technique that allows amplification of two or more products in parallel in a single reaction tube. This technology was first described by Chamberlain et al., in 1988, and has since been applied in diverse research areas, including analyses of deletions, SNPs, mutations, microsatellites, polymorphisms, pathogen identification, linkage analysis, gene expression, and forensic studies (Table).
Multiplex PCR, if sensitive enough, can be used to amplify and detect a single copy of a nucleic acid sequence and may also be used for both end-point and real-time PCR applications.
Multiplex PCR ensures standardization in certain experiments because identical reaction conditions and template amounts are used, pipetting and cycling condition variations are eliminated, and reliable comparison of results from a large number of fragments is achieved.
It also saves time and reagents for researchers performing large numbers of PCRs, hence its wide usage in various genotyping applications. However, certain challenges need to be addressed before the full potential of multiplex PCR can be explored.
Amplification of low copy-number target sequences in parallel with more abundant sequences is often limited by the generation of nonspecific PCR products and primer–dimers, and by the amount and quality of template DNA.
The establishment of optimal PCR parameters is the main factor influencing the success or failure of multiplex PCR. Traditionally, multiplex PCR has required extensive optimization of annealing conditions, enzyme amount, primer and probe concentrations, and buffer composition for maximum amplification efficiency of each target, leading to increases in costs and analysis time.
Compared with standard PCR systems using only two primers, an additional challenge of multiplex PCR is the varying hybridization kinetics of different primer pairs. Competition for reagent resources and the resulting artifacts can be potential problems in multiplex PCR.
Primers that bind with high efficiency could utilize more of the PCR reaction components, thereby reducing the yield of other PCR products. This often leads to unamplified DNA sequences and the absence of expected PCR products, eventually leading to false-positive or false-negative results.
In addition, traditional separation technologies (such as agarose slab gels) often do not provide sufficient resolution for post-multiplex PCR analysis and are not always optimal for separation of amplicons that differ by just a few base pairs.
Not addressing these bottlenecks leads to poor sensitivity, nonspecific amplification, and biased amplification of selected targets—challenges that scientists often encounter when initially setting up a multiplex PCR system, which involves several rounds of optimization with unpredictable success.
Increasing Sensitivity and Specificity
Several developments have been made in recent years to improve reaction specificity and sensitivity in multiplex PCR. Some studies require analysis of a large number of different mutations of a certain gene related to a disease (e.g., deletions or translocations) or other genetic information from an organism such as microsatellites. Including the necessary internal controls, a large number of PCR reactions are required when performing singleplex or lowplex-grade PCR analysis, leading to increases in both costs and analysis time.
Mutation analysis requires highly specific and sensitive detection of the mutated locus and a comparison with the wild-type locus. Additionally, it is useful to be able to detect the mutated and the wild-type locus in parallel or to be able to detect several different mutations in the same context simultaneously.
QIAGEN’s multiplex PCR technology has been specifically developed to overcome the challenges associated with multiplex PCR in both end-point and real-time PCR applications.
The newly developed QIAGEN Multiplex PCR Plus Kit affords highly sensitive multiplex PCR for advanced genotyping applications. It combines the benefits of a highly specific hot-start enzyme with a novel PCR buffer, which contains a balanced combination of salts and unique PCR additives. Together, these components ensure comparable efficiencies for annealing and extension of all primers in the reaction (Figure 1).
The increased hybridization efficiency and primer stability leads to maximal yields of specific PCR products—even for primer pairs that bind suboptimally to their target sequence under the chosen conditions. Consequently, most primer–template systems can be combined in a single reaction without optimization.
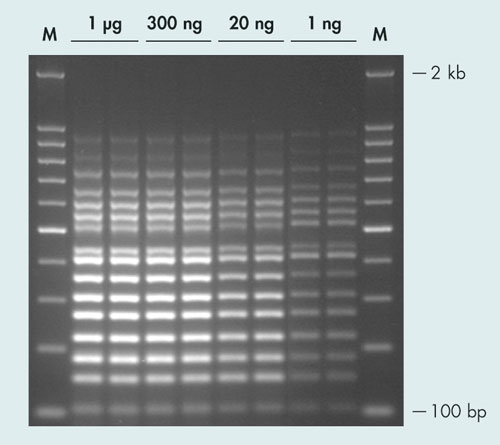
Figure 1. Successful 16-plex PCR over a wide range of template amounts: The QIAGEN Multiplex PCR Plus Kit was used to amplify 16 targets (99–955 bp) from various amounts of human genomic DNA, ranging from 1 ng to 1 µg. Successful multiplex PCR results were achieved in each case. M: GelPilot® 100 bp Plus Ladder.
One application of multiplexing PCR reactions is the analysis of microsatellites or minisatellites such as STRs or VNTRs, which are used in relationship studies or population genetics. The Type-it® Microsatellite PCR Kit, which is also based on multiplex technology, has been specifically developed and functionally validated for such analyses (Figure 2).
Microsatellite analysis on high-resolution detection systems such as capillary sequencers is often associated with challenges such as uneven product yield and substantial differences in the intensity of fluorescent signals. The Type-it Microsatellite PCR Kit, with its unique chemistry and optimized protocols, overcomes this limitation and ensures high product yields for all amplicons in a multiplex experiment.
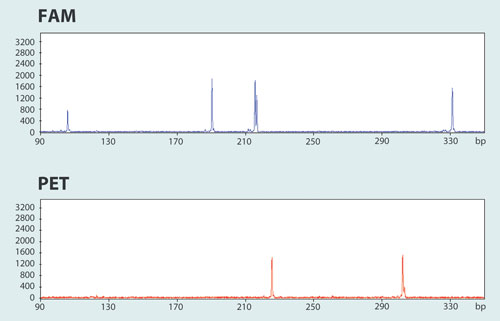
Figure 2. Optimization-free and reliable 13-plex STR analysis using the Type-it Microsatellite PCR Kit: Only 2 out of 4 channels (FAM™ and PET®) of a 3730 xl Capillary Sequencer (ABI), representing 6 of 13 analyzed STR loci, are shown. Using the Type-it Microsatellite PCR Kit under standard conditions, all 6 STR loci are reliably amplified.
Designing and Developing Successful Multiplex PCR Assays
Another prerequisite for successful multiplex PCR is the design of optimal primer pairs. The functionality and specificity of all primer pairs should be tested in single reactions before combining them in a multiplex PCR assay. The probability that a primer has more than one specific binding site within a genome is significantly lower for longer primers.
In addition, longer primers allow annealing at slightly higher temperatures where Taq DNA polymerase activity is higher. Problems encountered in multiplex PCR are frequently due to the use of incorrect primer concentrations or low-quality primers. QIAGEN PCR kits include streamlined protocols and preoptimized recommendations for primer concentrations and cycling conditions, ensuring multiplex PCR success.
Multiplex loci need to be selected based on the type of analysis that is being performed. For example, for transgene detection, two primer pairs can be used, one pair specific for the introduced transgene and the other pair specific for a wild-type DNA sequence. This second primer pair acts as a control for the amount and quality of the template DNA and cDNA.
This type of simple multiplex PCR analysis is often used in duplex real-time RT-PCR experiments where constitutively expressed genes such as the GAPDH gene serve as a control for measuring the abundance of a specific target gene. For complex multiplex assays with a variety of amplicons, where amplicons distinguish similar templates, such as those from related bacteria strains, primers should be located in regions that are not extremely variable.
In addition to the sequence of primers, the length of the generated PCR products should also be considered. Amplicon sizes must differ sufficiently in order to distinguish them from one another using agarose or polyacrylamide gels or capillary electrophoresis systems such as the QIAxcel® Advanced System, which ensures high-resolution separation of DNA fragments down to 3–5 bp.
Conclusions
Despite its technical challenges, multiplex PCR offers several advantages over singleplex PCR, such as simultaneous evaluation of template quantity and quality, inclusion of internal controls, and savings of time, costs, and reagents.
Due to its flexibility in assay design, multiplex PCR demonstrates significant benefits in an increasing number of applications—from gene analysis and linkage studies to human identification.(Table)
Exploiting the full potential of multiplex PCR in the future would involve maximizing the number of regions that may be simultaneously amplified. This is especially useful for studying genetic diseases associated with loci without apparent mutation hot spots or those harboring new or previously unidentified mutations.
Ulla Deutsch, Ph.D. ([email protected]), is associate global business director amplification, and Devika Mathur is technical and marketing writer at QIAGEN.

