
Study designs in proteomics aim for inclusivity—to characterize every protein in a sample. However, extracting proteins from complex biological samples involves the use of harsh buffers containing detergents and salts that are incompatible with the proteomics workflow. More proteins are lost in attempts to remove contaminants and interfering reagents from samples, resulting in increased variability and decreased reproducibility of data.


Rahul Samant, PhD, a scientist in the signaling program at the Babraham Institute in Cambridge, is the senior author of the study (left). Harvey Johnston, PhD, postdoctoral researcher in Samant’s group, is the lead author of the study (right).
Scientists at the Babraham Institute have upgraded conventional sample preparation protocols in proteomics by developing a new technique that increases the inclusion of hard-to-capture cellular proteins, thereby improving proteomics readouts. The investigators increased the recovery of less soluble proteins that contribute to the accumulation of toxic misfolded proteins in older cells. The new method also recovered more membrane-spanning proteins. This could improve the characterization of diagnostic and immunotherapy targets, and offer insights into how cells interact with their environment.
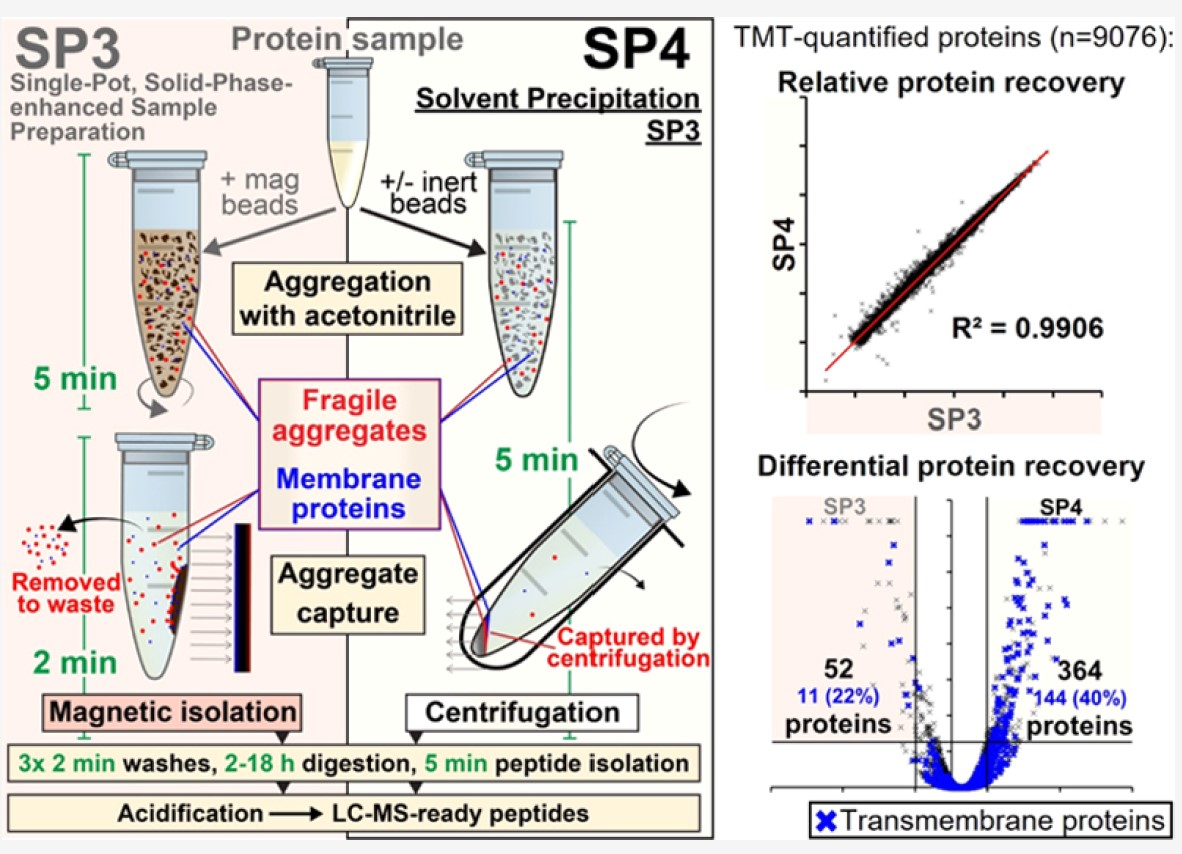
The technical advancement builds on an existing method of sample preparation called SP3. This current gold standard is a single-pot, solid-phase-enhanced sample preparation technique. It uses organic solvents and magnetic beads to denature and capture protein aggregates. This is followed up with washes to remove contaminants. Success in SP3 depends upon securely attaching proteins on magnetic beads. The method risks losing proteins that do not completely attach to the beads during washes. The loss and consequent processing costs multiply when samples contain higher concentrations of proteins or more hydrophobic proteins.
“Complete, reproducible extraction of protein material is essential for comprehensive and unbiased proteome analyses,” the authors noted.
The current method precludes the need to use magnetic beads, although the optional use of inert glass beads could simplify sample handling. Glass beads are available at 1/1000th the cost of magnetic beads and offers a low-cost alternative. The enhanced method replaces magnetism-based separation with centrifugation to identify more proteins with greater consistency.
“We omit magnetic beads entirely and instead employ acetonitrile-induced protein precipitation and centrifugation for protein capture and isolation—either bead-free, or with low-cost, inert glass beads,” the authors noted. “We name this method SP4.”
In test runs of SP4, the authors could recover protein yields that were equivalent or higher for 1–5000 microgram samples, with greater reproducibility, compared to SP3. Three other labs across eight sample types and five lysis buffers confirmed that SP4’s performance was equivalent or higher than SP3 in proteome characterization.
“SP4 offers a minimalistic approach to protein clean-up that provides cost-effective input scalability, the option to omit beads entirely, and suggests important considerations for SP3 applications all while retaining the speed and compatibility of SP3,” the authors claimed.
“Automated and efficient sample preparation is an essential pillar supporting increased throughput of proteomic analyses. SP4 looks like a useful addition to the sample preparation toolkit,” said Nikolai Slavov, PhD, an associate professor of bioengineering and the director of the single-cell proteomics center at Northeastern University College of Engineering. (Slavov was not involved in this study.)
The simplicity of the new method promises to make sample preparation in proteomics cheaper and more accessible. This will greatly support proteomics-based experiments in life science laboratories with low to medium budgets. In future experiments, the authors intend to apply the new method to investigate the mechanisms underlying the aggregation and elimination of misfolded proteins in cells.
References
Johnston H.E., Yadav K., Kirkpatrick J.M. et al. Solvent Precipitation SP3 (SP4) Enhances Recovery for Proteomics Sample Preparation without Magnetic Beads. Analytical Chemistry (2022).

