
January 15, 2008 (Vol. 28, No. 2)
Target Tissue Approach Can Be Used to Define Protein Networks and Treatment Modalities
Drug discovery and development are typically performed in the context of well-characterized model systems such as cell cultures and animals. Screening in such systems is likely to bias results given that the systems do not reflect the environment in which the drug will ultimately be used.
Human diseases are defined in a great many cases by cellular and, increasingly, molecular pathology. Of course, pathology defines normal biological states as well, which serves as a baseline for diagnosis and may be used to guide drug target discovery, and later on clinical treatment, in the context of the disease in the tissue microenvironment (TME).
The mainstays of molecular pathology such as immunohistochemistry (IHC) and fluorescent in-situ hybridization (FISH) are sensitive and capable of subcellular resolution. They are, however, not capable of the massively-multiplex assays routinely performed by mass spectrometry. Nor are they amenable to drug target discovery, in that the assay target must be known a priori and, in the case of IHC, have a validated, paraffin-compatible antibody. Such limitations have largely restricted these techniques to follow-on, validation roles in experimental workflows.
Two factors have inhibited omics-type investigations in samples traditionally used by and accessible to pathologists: formalin fixation of the sample and heterogeneity of the TME. Formalin fixation massively crosslinks cellular components making retrieval of individual molecular analytes essentially impossible.
Applications that use only short segments of a molecule existing between crosslinks can be performed however, witness IHC and FISH. The development of antigen retrieval in 1991 served to dramatically increase IHC sensitivity by partially reversing formaldehyde crosslinks, thereby making more of the protein retrievable. The same method significantly improves the quality of shotgun-proteomic analyses.
TME heterogeneity has similarly confounded proteomic analysis of tissue (Figure 1). To date, investigators have been confronted with two choices: analyze whole tissue or procure homogenous cellular populations from the TME via laser microdissection.
Analysis of whole tissue has not been palatable for the obvious reason that a multiplicity of cell types are typically present in the amount of tissue necessary for conventional proteomic analyses.
Therefore, the result will simply be an average of the proteomic states of the combined cell types. Comparison of such results across disease cases, in which the TME is often highly heterogeneous and often contains necrotic cells as well as vascular and inflammatory infiltration, is unlikely to result in meaningful insight into the disease system under study.
Alternatively, laser microdissection may be used to procure homogenous populations of cells, allowing for contextual analysis. The amount of protein obtainable, however, is typically well short of the quantity required for conventional shotgun-proteomic analyses. This has limited investigations using microdissected tissues to single dimension analysis by LC/MS, thus limiting proteome coverage, particularly of disease-relevant, low-abundance proteins.
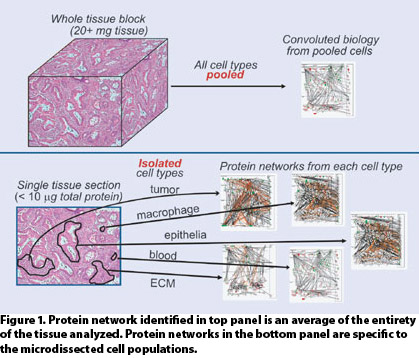
Figure 1
Gemini Platform
Calibrant (www.calibrant.com) developed the Gemini™ platform to enable high-resolution, 2-D shotgun-proteomic analysis of laser-capture microdissected populations of cells from formalin-fixed tissue sections. The basis of the platform is a first-dimension separation that utilizes transient capillary isotachophoresis/capillary zone electrophoresis (CITP/CZE) to resolve complex peptide mixtures retrieved from trace populations of laser microdissected tissue. This is coupled to nanoLC/MS.
Dissections of approximately 10,000 cells routinely yielded over 4,000 high-confidence quantitative protein identifications. This permits comparative proteomics across disease states as well as during drug development and treatment (Figures 2 and 3).
The capability to achieve comprehensive and comparative profiling from limited, targeted tissue samples can be attributed to the high-resolving power of CITP/CZE, which routinely separates peptides such that greater than 90–95% are found in only one or two fractions of a typical 30-fraction analysis. This acts to minimize analyte dilution across multiple fractions, which is critical in dealing with such limited sample amounts.
The high-resolution separation also serves to minimize matrix effects, which limit the dynamic range of the analysis, most notably at the electrospray source and in the mass spectrometer. Membrane protein detection, at roughly 30% of protein identifications, is consistent with what is predicted by gene encodings.
The proteome coverage enabled by Gemini permits the detection of low-abundance proteins including transcription factors and signaling molecules such as plasma membrane kinases. Canonical protein pathway coverage is also high.
Most significant, however, is the platform’s ability to define protein interaction networks in a pathological context. These networks are defined on the basis of proteins identified and known interactions culled from the literature. Because the comparative analysis is defined on the basis of the cellular or molecular pathology of the tissue under study, it is usually possible to profile normal tissue within the same case.
This establishes a calibration point from which to assess the diseased cells. Protein networks, which are differentially regulated between normal and disease states, enable both a highly sensitive and specific characterization of the molecular pathology as well as aid in a deeper biological understanding of its causative mechanisms and thus target selection.
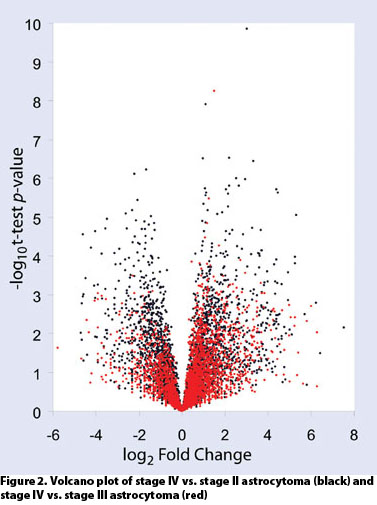
Figure 2
Protein Networks
A protein network, by virtue of the measurement of many proteins within the network, is a far more sensitive and reliable measure of disease status such as progression or response to therapy than protein markers used in isolation.
A network is typically composed of protein actors throughout the subcellular environment. This assists in target selection for differing objectives, for example, secreted and plasma membrane proteins as potential biomarkers in biofluids, plasma membrane receptor kinases as potential small-molecule and mAb targets, and cytoplasmic and nuclear proteins as potential small-molecule targets.
These networks may then be used going forward in validation and preclinical studies to guide target selection and evaluate drug efficacy. That is, is the network responding to treatment in a way that brings it closer to the state defined in normal tissue? This is where the network demonstrates its worth.
A single protein marker used for such evaluations would not be indicative of events occurring in the cell that bypassed that marker via an alternative pathway. Such an event is detectable in a network-based model because the downstream proteins are known and their response is measured. It is possible that the cell could use an entirely different pathway to circumvent the applied therapy. This too will likely be detected as a new change in a different network as a result of Gemini’s global proteome profiling capability.
It is often the case that the biology of model systems is substantially different from the biology encountered in human disease. Utilizing human tissue as a starting point to define disease biology allows for a more nuanced understanding of disease progression and the effect of therapy in the context of model systems.
Evaluation of potential therapies is ideally performed in a primary cell culture model because it is most biologically similar to the original disease. Utilizing Gemini in this context allows validation of the primary cell culture model—are the disease characteristic network modalities preserved? If so, Gemini may be used to systematically and globally assay a drug’s function and activity in the primary cell culture model.
Moving from preclinical to clinical trials, access to tissue in early-stage trials, in the absence of suitable biomarker profiles detectable in biofluids, becomes critical for confidently evaluating drug efficacy. Gemini can validate, pretreatment, that selected patients exhibit the dysregulated protein network upon which the trial therapy was developed. Post-treatment, Gemini can evaluate response to therapy. Gemini’s high sensitivity permits comprehensive, comparative analysis even from the increasingly small amounts of tissue procured via biopsy.
It is more and more evident that patients are willing to undergo biopsy if the sample may be used to guide critical clinical decisions such as switching treatment modalities when response to the standard of care is not indicated. This is equally true in the clinical trial arena where patients would, if possible, prefer to iterate through available treatment options until one is found that is effective for their disease type.
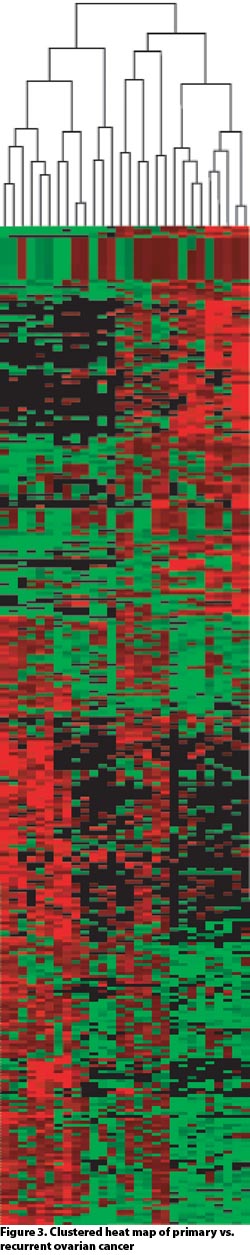
Figure 3
Brian M. Balgley, Ph.D., is CTO, and Cheng S. Lee, Ph.D., is CEO of Calibrant. Web: www.calibrant.com. E-mail: [email protected].

