September 15, 2006 (Vol. 26, No. 16)
Using SNP-CGH to Profile for Amplifications, Duplications, and Deletions
The beginnings of personalized medicine have been forged by recent advances in SNP genotyping technologies. It is now possible to accurately analyze over 600,000 SNPs across large patient sets, enabling powerful whole genome association studies. A new application of whole genome genotyping (WGG) is to profile, at increased resolution, the genome for DNA copy number, loss of heterozygosity (LOH), copy number polymorphisms, and other chromosomal aberrations routinely found in cancers and congenital disorders.
Karyotyping, fluorescence in situ hybridization, comparative genomic hybridization (CGH), and LOH analysis have been the standards for investigating rearrangements in genomic DNA and changes in DNA copy number. CGH has been used to identify homozygous deletions and amplifications, while LOH analyses have been used to investigate hemizygous deletions and copy-neutral LOH. LOH analysis was the foundation for the discovery of the well-known tumor suppressor genes RB1, WT1, and TP53. These classical methods are limited by their resolution and inability to assess genetic information.
Characterizing both changes in DNA copy number and allelic composition is crucial in understanding tumorigenesis. Therefore, the ability to collect both types of information at high resolution is of paramount importance.
Developed by Illumina (www.illumina.com), the Infinium™ WGG assay allows for the high-resolution profiling of both LOH and DNA copy number changes. Termed SNP-CGH, this technology can be used to profile the genome for amplifications, duplications, deletions, and LOH.
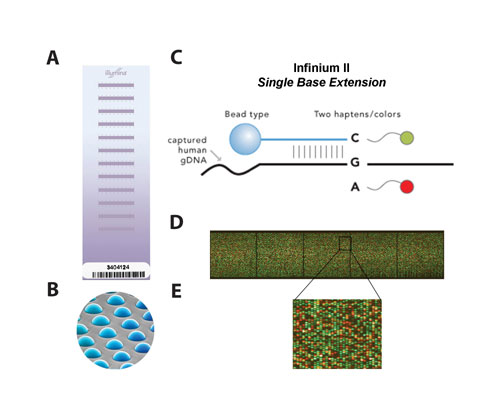
Figure 1Infinium II Single Base Extension
WGG BeadChip Content
Illumina manufactures several high density tag SNP genotyping arrays: the Sentrix® HumanHap300 (317k tag SNPs), the HumanHap550 (550k tag SNPs), and the HumanHap650Y (650k tag SNPs enriched in Yoruba tag SNPs). BeadChips are constructed by random assembly of bead pools into micro-well patterned stripes on a silicon substrate (Figure 1a, 1b).
Each stripe is loaded with a unique bead pool composed of tens of thousands of different bead types for a total complexity of hundreds of thousands of bead types across the BeadChip. Each bead type is immobilized with a decoding sequence and a locus-specific 50-mer oligonucleotide probe. The identity is determined by a hybridization-based decoding procedure. These BeadChips utilize the single-base extension (SBE) Infinium II assay to genotype tag SNPs selected from Phase I and II of the HapMap project. The median SNP spacing on the HumanHap300 is 5 kb and the HumanHap550 is 2.8 kb, enabling an effective resolution of ~50 kb and ~28 kb (10 SNP smoothing), respectively.
Infinium II WGG Assay Protocol
The Infinium II WGG procedure takes three days to complete and has four automated steps that are all performed on a Tecan robot system: (1) whole genome amplification, (2) hybridization to an oligonucleotide probe array (performed offline in a hybridization oven), (3) array-based SNP scoring assay, and (4) signal amplification. For the Infinium II assay, one bead type is used per SNP and the alleles are scored by SBE using differentially-labeled terminators (Figure 1c).
BeadChip Imaging
After completion of the assay, the BeadChips are scanned with a two-color confocal Illumina BeadArray™ Reader at a 0.84–1.0-µm pixel resolution. Image intensities are extracted and genotypes are determined using Illumina’s BeadStudio software. Aberrations were detected by visualizing SNP-CGH data in the Illumina Genome Viewer and Chromosome Browser.
Analyzing SNP-CGH Data
Genotyping data consists of two channel intensity data corresponding to the two alleles. Data can be plotted as rectangular coordinates of raw A versus raw B allele intensities (Figure 2a). Data are then normalized using a proprietary algorithm (Figure 2b), and genotype clusters are created with GenTrain software by clustering on ~120 normal individuals. Finally, the data are converted to polar coordinates, R and Theta (Figure 2c). Allelic intensities are transformed to allele frequency (allelic composition) using linear interpolation of the canonical clusters.
The comparison of normalized intensities between a reference and subject sample is the foundation of traditional array-CGH. SNP-CGH is different in that a combination of two genotyping parameters are analyzed: normalized intensity measurement and allelic ratio. Collectively, these parameters provide a more sensitive and precise profile of chromosomal aberrations. SNP-CGH also provides genetic information (haplotypes) of the locus undergoing aberration. Importantly, SNP-CGH has the capability of identifying copy-neutral LOH events, such as gene conversion, which cannot be detected with array-CGH.
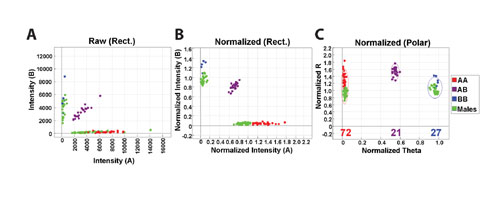
Figure 2: The encircled data points indicate the canonical genotyping clusters. Males are in green, and females are shown in other colors.
Detecting Chromosomal Aberrations
Aberrations are detected by comparing the normalized intensity of a subject sample to a reference sample. Illumina has developed two modes of SNP-CGH analysis. The first is a single sample mode in which reference values are derived from canonical genotyping clusters created from clustering on normal reference samples. The second is a paired sample mode in which direct intensity comparisons between a subject sample and its corresponding matched pair are performed.
Genomic plots of the log2 (Rsubject/Rreference), called log R ratio, and the allele frequency (AF) parameters along the chromosome form the basis for detecting chromosomal aberrations (Figure 3).
Allelic Imbalance in Cell Lines
The ability of the HumanHap550 genotyping BeadChip to detect chromosomal aberrations in the cancer cell lines HL60 (promyelocytic leukemia) and MCF7 (breast adenocarcinoma) are demonstrated in Figure 3. In HL60, deletions of varying size on chromosome 9—a ~20-Mb deletion on the p arm and a ~1.8-Mb deletion on the q arm—are shown in Figure 3a. These hemizygous deletions, from two copies to one copy, are manifest as a downward deflection in the log R ratio and a loss of heterozygotes in the AF.
The LOH+ module of BeadStudio allows for the user to annotate regions of interest, shown by the various bookmarks superimposed on the profiles. Other examples of aberrations in HL60 include monoallelic duplication of chromosome 18 as indicated by an increase in the log R ratio and split of the heterozygotes into two states: one at 0.67 (2:1 ratio) AF and another at 0.33 (1:2 ratio) AF (Figure 3b). Figure 3c demonstrates several small amplifications in HL60 on chromosome 8. Two adjacent homozygous and hemizygous deletions are observed in MCF7 (Figure 3d).
The gene annotation provided in the LOH+ module allows users to quickly analyze affected gene regions, and in this case, both copies of MTAP are homozygously deleted, whereas CDKN2A and CDKN2B are hemizygously deleted.
SNP-CGH provides a rapid means for simultaneous genomic profiling of physical and genetic anomalies. The foremost advantage of Illumina’s WGG BeadChips for SNP-CGH is that these arrays are easily manufactured and possess intrinsic scalability. The feature density is now greater than 550,000 SNPs across the human genome.
The Infinium assay enables both unlimited multiplexing and relatively unconstrained SNP selection, allowing development of high density tag SNP arrays. Additionally, automation of the Infinium assay allows large numbers of samples to be robustly processed and analyzed. The combined measurement of both allelic ratios and normalized intensities provides enhanced detectability of aberrations and allows identification of copy-neutral genetic anomalies, such as uniparental disomy and mitotic recombination. SNP-CGH can also collect allelic information on deletions, duplications, and amplifications, which has significance to cancer therapeutics.
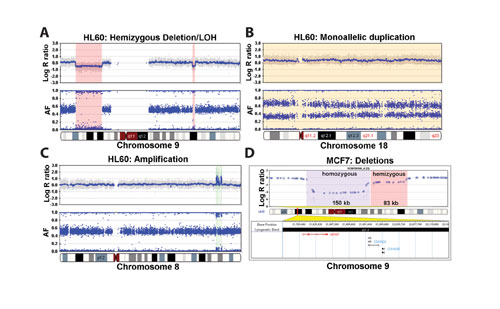
Figure 3: Genomic profiles of cancer cells on the HumanHap550.
Kevin L. Gunderson, Ph.D., is a principal scientist and Daniel A. Peiffer, Ph.D., is product manager at Illumina. Web: www.illumina.com. Phone: (858) 202-4500. E-mail: [email protected].

