
January 15, 2011 (Vol. 31, No. 2)
LC-MS/MS Allows for Accurate and Sensitive HT Quantification
Antisense oligonucleotides, siRNAs, miRNAs, aptamers, and other small nucleic acids are gaining prominence in the biopharmaceutical pipeline. The need for robust, accurate, and efficient ways of detecting such oligonucleotides and their metabolites in complex bodily fluids is crucially important for pharmacokinetic, pharmacodynamic, and exposure-response studies, as well as for the future of patient care.
There are several ways in which these measurements are accomplished, including by LC-MS/MS, HPLC-UV, capillary gel electrophoresis, and immunologically based approaches. Each of these methods offers certain advantages and limitations. The LC-MS/MS approach is sensitive and specific, accurate and reproducible, and is amenable to fast, high-throughput analysis. The Table summarizes some of these benefits.
An LC-MS/MS assay offers three levels of discrimination. First, samples are separated based on their mobility through the chromatography column—generally based on some combination of size, polarity, charge, and, perhaps, affinity. A typical chromatograph will indicate the amount of substance (detected by UV absorption, MS signal intensity, etc.) versus the time (or fraction number) at which it was eluted.
When a triple quadrupole mass spectrometer is used, the eluate is ionized as it enters the mass spectrometer, where it is scanned for mass/charge ratio (m/z) in Q1 based on its molecular weight and charge states. Selected ions with specific m/z from Q1 are then allowed to collide in Q2 with gas molecules, fragmenting them into pieces, which are then measured for m/z by a second detector (Q3).

On the basis of multiple specific MRM (multiple reaction monitoring) “transitions” from a compound to unique fragments, the instrument is able to discriminate among starting compounds that could not otherwise be differentiated. This is especially true for poly-ionic oligonucleotide compounds that usually have multiple unique ionic charge states in Q1 scan (Figure 1).
Scientists at Tandem Labs have developed and validated dozens of sensitive, high-throughput LC-MS/MS assays allowing us to quantify oligonucleotide molecules in bodily fluids such as urine and plasma. This tutorial presents an outline of the development of a LC-MS/MS method and an example of its use: the detection and quantification over a broad dynamic range of a model compound from human plasma.
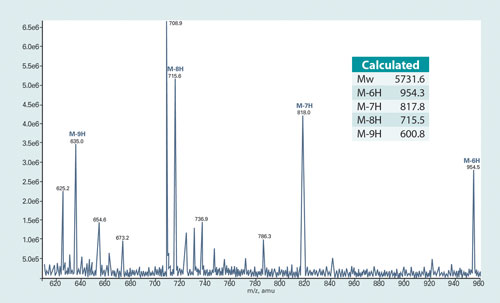
Figure 1. Q1 scan mass spectrum of TL0901
Optimizing LC-MS/MS Conditions
We are interested in accurately quantifying small oligonucleotide therapeutics in bodily fluids such as plasma. Here we randomly chose an 18-mer phosphorothioate oligonucleotide TL0901, with molecular weight of 5731.6, as a model compound. As mock metabolites we created shortened versions of TL0901—TL0901 (n-1) is the corresponding 17-mer, TL0901 (n-2) the corresponding 16-mer, and so forth.
Using ion-pair reverse-phase HPLC, TL0901 could be fully separated chromatographically from its n-6 metabolite (12-mer), but the chromatogram shows TL0901’s peak retention time slightly overlapping with that of the n-4 metabolite (14-mer).
Thus HPLC alone is inadequate when separating this oligo from its longer metabolites. As such the selection of an appropriate MRM transition for this oligonucleotide is needed to distinguish it from these metabolites. This can be done by analyzing corresponding neat oligonucleotide solutions on a mass spectrometer equipped with an appropriate HPLC system under optimized LC conditions.
Figure 1 shows a typical Q1 scan mass spectrum of TL0901 on an API5000 with a turbo ion spray ion source. The m/z of the various peaks allows the ionization state of the compound to be calculated based on its molecular weight. For example, the peak at 715.6 is TL0901 losing eight protons (M-8H), while that at 818.0 is TL0901 losing seven protons (M-7H).
From Q1 the ionized compounds undergo fragmentation in Q2, yielding a variety of smaller products, and the fragments are then analyzed in Q3. Many of these fragments (for example, that with m/z of 319) are seen to be shared among the products of the different parent ions (for example, M-8H and M-7H). Therefore, multiple signature MRM transitions can be used for monitoring TL0901.
For quantitative determination of TL0901 in pure neat solution samples, any of these signature MRM transitions can be used. However, for complex samples that contain metabolites and components from a biological matrix, the key, then, is to choose which transition(s) will accurately quantify full-length TL0901. It is especially important to ensure that the chosen transition does not suffer interferences from the compound’s metabolites that have retention times overlapping with those of the parent compound.
Figure 2. Chromatogram of TL0901 (n-1) and TL0901 (n-2) (top trace), and their interferences to TL0901 transitions (lower traces)
Figure 2 is an example of the process of selecting the appropriate MRM transitions. We injected a neat solution containing n-1 and n-2 metabolite mixture (2,500 ng/mL, the highest testing concentration) onto a LC column under optimized conditions (column: 2×50 mm C18 column; mobile phases: HFIP and TEA buffered water and methanol).
In addition to monitoring the corresponding n-1 and n-2 metabolite MRM transitions, we also monitor up to 10 different MRM transitions identified for TL0901. The top trace of Figure 2 shows the chromatogram of TL0901 (n-1) and TL0901 (n-2). The bottom three traces show the chromatograms monitored with three MRM transitions of TL0901.
The expected peak retention time of TL0901 is approximately 1.32’ in this experimental condition. The major peaks in both the 645.9 → 319 and the 817.7 → 319 transition traces represent the contribution from TL0901 (n-1) and TL0901 (n-2), making them inappropriate for quantitative analysis of TL0901.
However, neither of these two metabolites contributes to a detectable peak of the 715.4 → 319 transition at the expected TL0901 retention time (~1.32′).Therefore, the quantitation of TL0901 will not be affected by the n-1 and n-2 metabolites by monitoring the 715.4 → 319 transition during LC-MS/MS analysis.
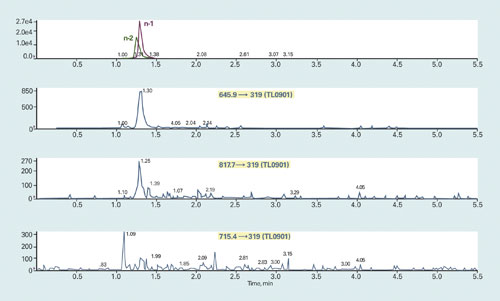
Figure 2. Chromatogram of TL0901 (n-1) and TL0901 (n-2) (top trace), and their interferences to TL0901 transitions (lower traces)
LC-MS/MS in Plasma
To analyze TL0901 in complex biological matrices by LC-MS/MS, samples must be precleaned or extracted together with an appropriate internal standard (a 12-mer analog in this case) prior to running. We have found both solid-phase extraction and liquid-liquid extraction (LLE) methods were effective for the extraction of phosphorothiate oligonucleotides. However, LLE produced better and more consistent recovery (>75%).
This method has been fully validated for quantitation of TL0901 in human plasma under good laboratory practice (GLP) conditions, including intra- and inter-day precision and accuracy, selectivity/specificity, metabolite interference, and various stability tests. The validated linear range is from 10.0 to 2,500 ng/mL; both inter-day and intra-day precisions were less than 6%, with a mean accuracy (%bias) of -10% to 1.0%. The assay matrix selectivity was also tested at lower level of quantification in 10 different lots of plasmas, and all stability tests are acceptable per the GLP guideline set forth by FDA.
LC-MS/MS provides an unambiguous, sensitive, and efficient means of detecting and quantifying oligonucleotides in complex biological fluids. We have validated many LC-MS/MS assays for various structured oligonucleotides in various matrices and analyzed thousands of samples in support of both clinical and nonclinical GLP studies around the world. We believe that LC-MS/MS represents the future high-throughput platform for the bioanalysis of oligonucleotide therapeutics.
Laixin Wang, Ph.D. ([email protected]), is a method development group leader at Tandem Labs, a LabCorp company.

