
Researchers at Cold Spring Harbor Laboratory have identified three different molecular subtypes of the progressive, fatal neurodegenerative disorder amyotrophic lateral sclerosis (ALS), and suggest the findings could help to direct the development of new treatment approaches for some patients. The team used machine learning algorithms to find patterns in the transcriptomes of post-mortem brain tissue samples from patients with ALS. The results showed that the majority of samples displayed hallmarks of oxidative and proteotoxic stress. A second group of samples showed predominant signatures of glial activation, but a fifth of the samples exhibited high expression levels of what are known as “jumping genes”—retrotransposons—and signatures of dysfunction in the gene TARDBP, which codes for TDP-43, a protein that has recently been linked with regulating retrotransposon activity. Retrotransposons are genetic elements that can randomly move between sites in the chromosome and alter gene expression.
“These jumping genes are telling us about patients who have TDP-43 pathology,” said associate professor Molly Gale Hammell, PhD. “We really don’t know why one patient would have one set of symptoms versus another, and we’re trying to answer that question.” Hammell’s team headed the reported research, which is described in Cell Reports in a paper titled, “Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia.” The Cold Spring Harbor scientists collaborated with researchers from Stony Brook University, Johns Hopkins University, Georgetown University Medical Center, the NYGC ALS Consortium, New York Genome Center, and University of California, San Diego.
There is no known cure for ALS, and only two FDA-approved treatments that appear to slightly slow disease progression, the authors explained. ALS is a largely sporadic disease with no known genetic mutation or family history. However, the team continued, “Large-scale patient sequencing studies have identified a growing number of genes in which mutations are linked to ALS.” And while mutations in TARDBP gene are rare in ALS, “sporadic ALS patients are known to show cytoplasmic accumulation and aggregation of TDP-43 protein (produced by the TARDBP gene) in the motor cortex and spinal cord, two tissues where motor neuron loss occurs … nearly all ALS patients exhibit cytoplasmic aggregates of TDP-43 in the affected tissues.”
Interestingly, while studies by Hammell’s lab and other researchers have shown that TDP-43 acts to block retrotransposon transcripts in animal models of TDP-43 pathology, a role for the protein in ALS hasn’t been uncovered. “… a role for TDP-43 in general retrotransposon silencing has not been demonstrated, nor whether TDP-43 pathology in ALS patients correlates with retrotransposon de-silencing.”
For their studies, the researchers analyzed data from the NYGC ALS Consortium collection, which includes deeply sequenced transcriptomes from the frontal cortex of 77 ALS patients, together with neurological and non-neurological controls. For some patients, samples had been taken from different areas of the frontal cortex, so Hammell and colleagues analyzed a total of 148 transcriptomes from ALS patients, and 28 from controls.
The researchers applied the machine learning algorithms to determine whether the ALS patient samples fell into different molecular subsets, according to their specific gene signatures. The results suggested that there were three molecular subtypes of ALS sample. Sixty percent of the samples were categorized as ALS-Ox, and demonstrated expression signatures consistent with oxidative and proteotoxic stress. This group of samples also showed elevated levels of genes that have previously been linked with ALS. A second ALS subgroup, comprising 19% of samples and dubbed ALS-Glial, was defined by increased expression of glial markers, and upregulation of innate immune pathways that are typically elevated in activated microglia. The remaining 20% of samples, categorized as ALS-TE (TE for transposable elements) demonstrated retrotransposon re-activation as a dominant feature. “The ALS-TE samples showed an enrichment for transposon expression as the most significantly enriched pathway relative to controls,” the authors stated.
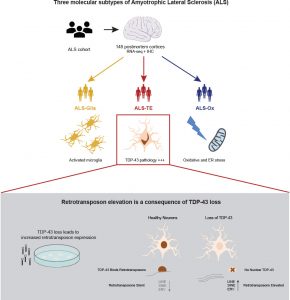
TDP-43 is one of the proteins that silence jumping genes. But when TDP-43 accumulates in clumps or aggregates in the nerve cells of ALS patients, it fails to silence the jumping genes. Hammell’s group found these telltale signs of TDP-43 pathology, or abnormal characteristics, and elevated transposons in the ALS-TE patient samples. The gene expression patterns indicated de-silencing of jumping genes for the subset of patients with the most extensive TDP-43 pathology. The authors suggest that their results “establish a correlation between TDP-43 dysfunction and transcriptional re-activation of retrotransposon sequences in ALS patients tissues.” However, the team further acknowledged, “the role of transposable element expression is a relatively new topic in the study of neurodegeneration. Moreover, the connection between TDP-43 and transposable elements has only recently been explored.”

“That’s one of the things we’re really excited about as a possibility,” Hammell added. “Going forward, we want to understand whether transposons are causing the disease, and whether they’re involved in other diseases that also tend to have TDP-43 aggregates, like frontotemporal dementia and some subset of Alzheimer’s.”
