
June 15, 2018 (Vol. 38, No. 12)
Sometimes Less Is More as Honing in on a Single Cell Grants New Insights
When cell populations are analyzed, genomic, transcriptomic, and proteomic intricacies tend to lurk in the shadows that gather whenever average measurements are taken. When individual cells are analyzed, however, these intricacies may be brought to light. Turning dark blurs into vibrant mosaics is the business of single-cell analysis—and business is good. Single-cell analysis is yielding intriguing insights into cell and system development, the physiology of aging, the noncoding genome, and the molecular drivers of cancer and other diseases.
Although still largely confined to a subset of devoted researchers, single-cell analysis is starting to enjoy wider use, growing as single-cell platforms become more accessible, as expertise is gained and disseminated, and as diverse methodologies gel.
With its emphasis on the individual, single-cell analysis is inherently democratic. How fitting, then, that single-cell analysis should show signs of being democratized. In fact, at a recent single-cell event, Single Cell Genomics 2017, the stated theme was, “Sometimes the sum of the parts is greater than the whole.” This event, which was held at the Weizmann Institute of Science, attracted single-cell pioneers and experts eager to share their insights, many of which are summarized in this article.
Signatures of Aging
Some of the presenters at Single Cell Genomics 2017 demonstrated how single-cell RNA sequencing data from primary cells could be used to illuminate genetic and transcriptional processes that occur in aging human tissue. For example, Stephen Quake, Ph.D., a professor of bioengineering at Stanford University and co-president of the Chan Zuckerberg BioHub, discussed his work with Seung K. Kim, Ph.D., a professor of developmental biology at Stanford. The Quake and Kim laboratories used single-cell transcriptomics to evaluate the effects of aging on the pancreas. In this work, 2544 human pancreas cells spanning six decades of age were analyzed.
Entire transcriptomes were compared. Findings showed that transcriptional entropy increased globally with age. Cells were diverging, transcriptionally. With age, the mutational load increased in ways that were characteristic of increasing oxidative damage. This mutational burden increase was unexpected and challenging to measure at the single-cell level.
Another all-Stanford collaboration, between Dr. Quake’s group and the laboratory of Liqun Luo, Ph.D., a professor of biology, aimed to understand if single-cell transcriptomic measurements really reflected physiological ground truth as cell biologists have defined it for decades in terms of measuring cellular identity. In this collaboration, the research focused on Drosophila olfactory projection neurons (PNs). These are among the best-characterized neuronal types.
Different PN classes target dendrites to distinct olfactory glomeruli. PNs occur in different clusters; cells of the same class exhibit identical anatomical and physiological properties. These physiologically distinct cell types, Dr. Quake indicated, were identified by analyzing transcriptomes. Also discovered were new transcription factors that are involved in the way the cells wire up together and decide which cells they are going to recognize.
“The first challenges in the single-cell genomics field were to develop the technical methods and prove they worked,” said Dr. Quake. “Now that that has been accomplished, new questions have emerged.” For example: What is the best science one can do with these approaches?
“We find that these technologies represent a valuable tool in the arsenal one brings to big biological questions,” Dr. Quake continued. “More and more we will see single-cell approaches used to complement conventional approaches to understanding how cells evolve and what their roles are in the human organism.”
Current methods for single-cell genomics include cell sorting, microfluidic methods incorporating fabricated valves, and microfluidic emulsion-based approaches. A new method, Microwell-seq, a high-throughput and low-cost scRNA-seq platform, was recently described by a group from Zhejiang University. Different methods present different strengths and are suited to the resolution of different of problems.
One Pot, Two “Omes”
Hundreds of transcripts from a single cell can be measured in high throughput. This kind of transcriptomic analysis could be complemented, scientists hope, by high-throughput multiplexed single-cell proteomic techniques. The difficulty, however, is that the number of proteins that can be measured per cell is limited using mass spectrometry.
Progress toward transcriptomic/proteomic complementarity has been reported by Savas Tay, Ph.D., an associate professor at the Institute of Molecular Engineering and the Institute for Genomics and System Biology at the University of Chicago. In Dr. Tay’s laboratory, a microfluidic single-cell technology is being developed that could potentially measure hundreds to thousands of individual proteins and complexes all in the same pot, along with the transcriptome.
Two antibodies tagged with short oligos of a known sequence target a protein. After the antibody binds to the proteins, cells are lysed, the oligos are sequenced, and the binding abundance in the cell is determined. This proximal ligation assay approach increases sensitivity and minimizes antibody noise and off-target effects.
Protein complexes can also be measured; 50 antibody-oligo pairs could measure up to 2500 complexes. The sequencing readout makes the technology compatible with RNA sequencing. In a single droplet, both the transcriptome and proteome are measured.
“In single-cell research, you want to measure as many species as possible,” noted Dr. Tay. “We would like to continue using droplet-based sequencing and develop protocols that will label the cells spatially and temporally to continue to use our high-throughput methods.”
Current technologies do not allow the measurement of temporal evolution of cells in a high-throughput multiplex fashion; endpoint measurements do not provide temporal information. Much effort is required to understand temporal aspects through static but still computationally heavy measurements.
Longitudinal cell-imaging experiments to study temporal and spatial patterns are a cornerstone of biology, but the low-resolution protocols are limited by pipetting requirements. A new high-throughput microfluidic culture and live-cell analysis system is under development to keep cells alive and expose them to reagents in a spatially and temporally controlled fashion to mimic the native environment. The device performs millions of pipetting steps in a fully automated fashion, and it has been used to understand neural stem cell development.
“We need to keep cells alive for dynamic high-throughput multiplexed measurements in a spatially resolved manner—ideally, in intact tissue to maintain the tissue microenvironment and context,” Dr. Tay commented. “Right now, the field is trying to inch forward with different approaches, but this is the nirvana of single-cell analysis and would be a quantum leap.”

Microfluidic single-cell technology is being developed that could potentially measure thousands of individual proteins and protein complexes along with the transcriptome.
Epimutation Rates
As cells divide and propagate, they acquire errors in their genetic information as well as their epigenetic information, a phenomenon termed epimutation. Earlier studies indicated that the average epimutation rate in cancer is significantly elevated as compared to normal tissue.
Additional studies are being conducted at the laboratory of Dan Landau, M.D., Ph.D., assistant professor of medicine at Weill Cornell Medicine and a core member of the New York Genome Center. Dr. Landau and colleagues are using high-throughput multimodality single-cell protocols and integrative analytics to capture the DNA methylome, the transcriptome, and the genotype of normal and malignant B cells.
Multimodality technologies were applied to patient samples with chronic lymphocytic leukemia along with normal B-cell samples to probe the average epimutation rate and the rate distribution within cells. The distribution in normal tissue was quite wide. In contrast, in leukemia samples the distribution was narrow with uniformly elevated rates. This observation corresponds to epimutation as a molecular clock.
Whereas leukemia cells come from a single clonal origin and therefore have a uniformly high number of divisions, normal B cells have different evolutionary histories; some have just emerged from the bone marrow, and others are older, reflected by a low or high epimutation rate, respectively.
“The epimutation information can also be used as a native lineage tracing system, [allowing investigators] to infer the phylogenetic relationships between individual cells,” explained Dr. Landau. “With this approach, high-resolution lineage trees are created directly from primary patient cancer samples.”
The topography features of the malignant lineage trees are consistent with clonal transformation, demonstrating strong drift early after transformation, and long branch lengths, a result of the high division rate in cancer. In contrast, normal B-cell lineages show deeper structures due to the differentiation of one cell type to another, with shorter branch lengths commensurate with lower numbers of divisions in their histories.
Since the methylome, transcriptome, and genotype of the single cells are captured, genotypic and transcriptional information can be projected on to the lineage tree, allowing clonal divergence to be defined as well as heritable transcriptional variation. This approach was also applied to study the evolutionary dynamics that occur as patients receive therapy.
Even in genetically homogeneous populations, cells with shared lineage histories could be identified that were differentially impacted by with therapy. The joint transcriptional information further allowed the study of the potential mechanism for the differential response to therapy among cells with different lineage histories within the same patient.
Dark Matter
“There is a good reason why we refer to the noncoding genome as dark matter,” commented Gary Hon, Ph.D., assistant professor of obstetrics and gynecology at UT Southwestern Medical Center. “We know it is there. We know it plays important roles in development and disease. But we have little clue what most of it does.
“Thanks to the efforts of consortia like ENCODE [the Encyclopedia of DNA Elements, a project that was launched by the National Human Genome Research Institute in 2003], a map of the noncoding genome and what it encodes now exists. But despite having a great map, the single greatest challenge is that the vast majority of the millions of noncoding elements have no known function.”
This is a big problem that Dr. Hon’s group approaches from a systems biology perspective. The group invests heavily in developing new biotechnological tools to functionally interrogate the noncoding genome using single-cell approaches.
According to Dr. Hon, the best way to understand the genome is to systematically perturb as much of it as possible. By directly measuring the functions of thousands of noncoding regions, Dr. Hon and colleagues mean to derive the fundamental principles that govern these regions.
Recently developed techniques, such as Perturb-Seq, CRISPR-Seq, CROP-Seq, and Mosaic-Seq, will play important roles. The key feature they share is the ability to detect CRISPR-mediated genetic perturbations in individual cells, as well as the phenotypic consequences of these perturbations. Together with recent developments enabling thousands, or even millions, of single cells to be individually sequenced, these approaches are scaling the functional perturbation of the genome, raising the technique to new heights.
In a single well-controlled experiment, Mosaic-Seq was used to directly measure the activities of 71 enhancers. Surprisingly, even though many of these regions are predicted to be strong enhancers based on certain epigenetic features, most enhancers were found to exhibit no functional activity when repressed alone.
Often, multiple enhancers had to be repressed to elicit a functional effect, suggesting that many enhancers are functionally redundant. This redundancy means that understanding enhancer function will likely require perturbing multiple enhancers in the same cell, and further underscores the importance of applying systematic single-cell techniques like Mosaic-Seq to study these regions.
Technological innovations have positioned the single-cell genomics field to make great strides in the next few years, enabling a deeper understanding how the dark matter of the human genome functions. Just as the ENCODE project systematically mapped millions of noncoding elements, there will be a rapid acceleration of efforts to functionally test these regions.
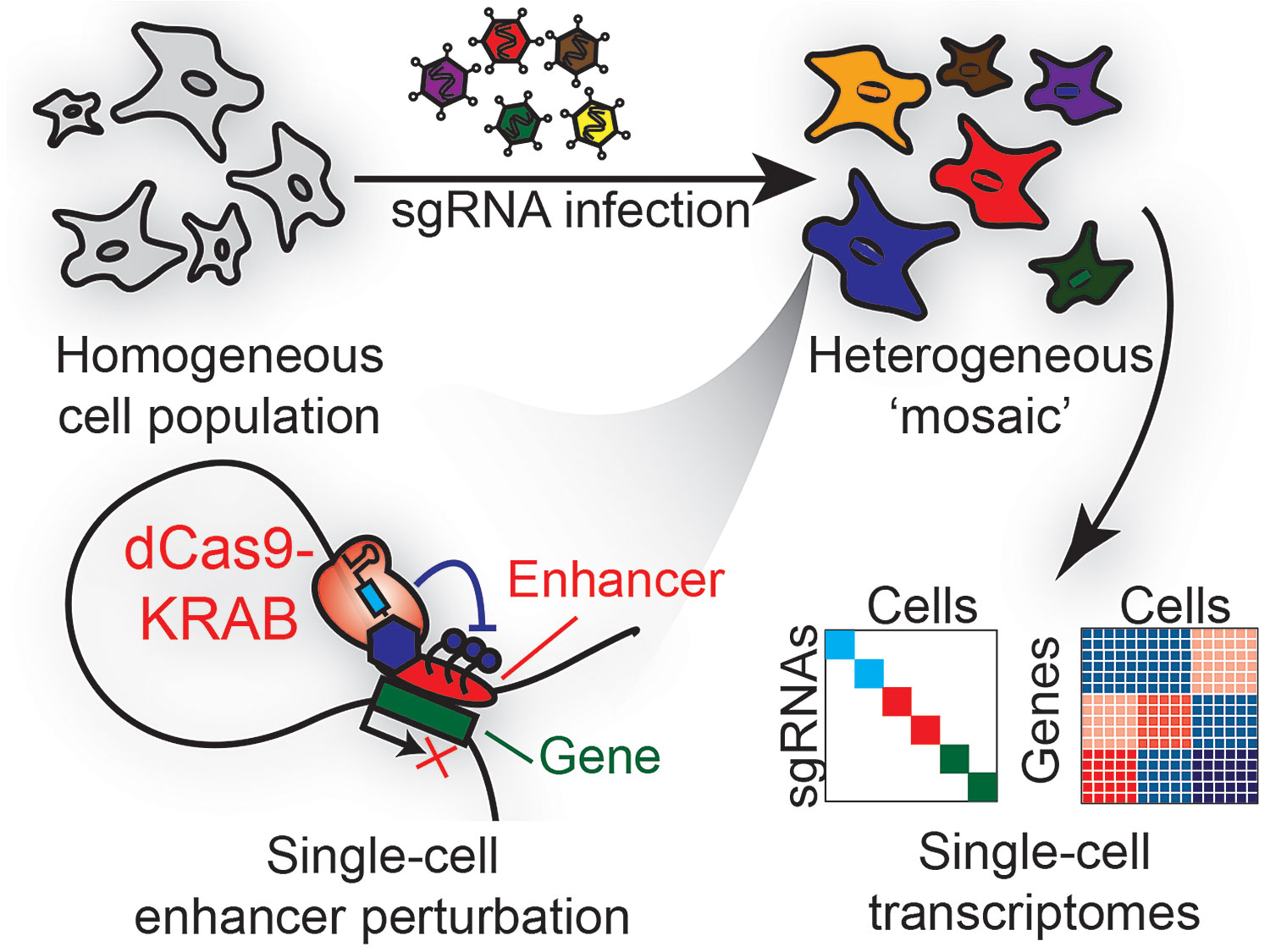
Mosaic-Seq was developed to systematically perturb enhancers and measure their endogenous activities at single-cell resolution. This high-throughput technique uses a CRISPR barcoding system to jointly measure a cell’s transcriptome and its sgRNA modulators, thus quantifying the effects of dCas9-KRAB-mediated enhancer repression in single cells.
High-Throughput Targeted Single-Cell DNA Analysis
Cancer is heterogeneous and understanding how multiple DNA mutations co-occur during disease progression is critical to better informing therapy. Current next generation sequencing approaches lack the high sensitivity and single-cell resolution to unambiguously identify rare subclones with co-occurring mutations necessary to fully understand the complexity of the disease, according to Dennis Eastburn, Ph.D., chief scientific officer and co-founder, Mission Bio.
Targeted single-cell DNA analysis offers the opportunity to resolve genetic heterogeneity, he explains. However, methods to characterize heterogeneity and mutational status at single-cell resolution have lagged behind RNA expression analysis due to the challenges of accessing and amplifying genomic DNA. Because single-nucleotide variants (SNVs) and indels are especially informative for selecting gene specific targeted therapies, it is essential to directly assess their mutational status within a cell.
“The Tapestri Platform for single-cell DNA analysis provides accurate and targeted identification of SNVs and indels to enable precise variant calling, thereby overcoming the incomplete genome coverage and artifactual mutations introduced from single-cell whole genome amplification approaches,” says Dr. Eastburn. “This targeted approach provides a robust and cost-effective solution for clinically relevant and actionable information.”
Work from leading cancer centers using this new precision genomics platform has found that subclones responsible for expanding and driving relapse can be found at very low levels, below 0.1 percent of the tumor population, prior to treatment, he continues.
“Significantly, these rare subclones were undetectable with bulk sequencing. This high-resolution view of clonal architecture has the potential to alter therapy selection and improve disease outcomes,” says Dr. Eastburn.
BD’s Single-Cell Analysis System
Massively parallel single-cell RNA sequencing is a powerful tool for examining individual differences between cells. Technologies used in single-cell research for gene expression have continued to improve dramatically in recent years—sequencing tens to hundreds of individual cells one-by-one used to be the only way to examine cellular heterogeneity—but new technologies that utilize unique molecular identifier labels. BD officials say the company’s RhapsodyTM single-cell analysis system can measure thousands of genes across tens of thousands of single cells in parallel.
“Despite these advances, the lack of protein expression information with single-cell RNA sequencing can make it challenging for researchers to identify cells that exhibit similar transcriptional profiles,” notes Stephen Kulisch, senior director, applied markets at BD. “They may have a low abundance of transcripts, splice variants or have data that doesn’t correlate well between protein and gene expression. Evaluating multiple modalities in single cells can illuminate critical differences and power an improved understanding of cellular composition and how cells function or complex diseases and conditions.”
Protein cell-surface identification can be added to RNA-seq single cell experiments through an oligo-conjugated antibody panel, such as BDTM AbSeq, according to Kulisch.
“This technology provides a more distinct and robust clustering of different cell subsets, enabling researchers to more deeply profile cells to further elucidate complex biological systems. It also provides one readout to analyze both protein and RNA, reducing experimentation time and increasing information received in a single run,” he adds Kulisch.
Recently, the BD team demonstrated the capabilities of this technology by examining T-cell subsets using BD AbSeq. By adding protein-marker measurement to high-throughput single-cell RNA sequencing technology, researchers were able to identify rare cell populations including a rare and recently described T-cell subset and stem memory T-cells.

