
Antibody discovery is riding a single-cell wave, allowing massive numbers of B (and T) cells to be individually interrogated for a variety of parameters while at the same time retaining their antigen receptor chain pairing. What might have taken half a year can now be accomplished in a day or a week, at a throughput that was unimaginable back when today’s principal investigators were graduate students. This was the focus of the Deep Sequencing and Single Cell Analysis for Antibody Discovery conference, held as part of last month’s PepTalk in San Diego.
Working in reverse
Vaccines against viruses are typically designed to mimic an infection in a way that’s safe for the healthy recipient—using an attenuated or killed virus, for example, or perhaps selected portions of the native virus—to elicit an immune response that will protect against a real challenge. But the typical approach doesn’t always work. An example of how it may fail was cited by Bryan Briney, PhD, assistant professor of immunology and microbiology at Scripps Research: “It has been tried for HIV, and it hasn’t worked.”
His group—part of a large collaboration—has been focused on an approach many call “reverse vaccinology.” The idea is to find the kind of antibody response you’re trying to elicit, and then work backward to a vaccine immunogen that will elicit it.
“The vaccine protein or set of proteins might not necessarily look like the virus very much,” Briney pointed out. The native (germline) antibodies must undergo many rounds of mutation and selection (termed affinity maturation) against HIV before they are able to bind and neutralize the virus. So much so, in fact, that mature antibodies that recognize HIV don’t bind the antigens that their unmutated precursors do, and vice versa.
The idea is to seek out the rare B cells that make the rare antibodies that have the potential to mature into broadly neutralizing antibodies and, as Briney explained, “kind of kick them off and get them pointed in the right direction.” That is, the B cells are subjected to the right subsequent vaccine boosts with either the same or a different antigen. “We can continue to shepherd their development toward the function we’d like them to have,” Briney added.
The process involves computationally inferring what the germline antibody sequences looked like, and engineering proteins using yeast display directed evolution approaches to find what they bind. “Once we find an immunogen that binds a lot of these inferred precursors,” Briney elaborated, “we can then take that immunogen and sort B cells from healthy, uninfected people, to see what kinds of things this immunogen binds and would presumably be stimulated in the vaccine approach.” When Briney and colleagues applied an iterative process of engineering and functional evaluation, they found, Briney recalled, immunogens that bind “orders of magnitude better than a native HIV envelope.”
Because an antibody’s specificity is dependent on the pairing of its unique heavy and light chains, in order to capture that native specificity, sequence information must come from individual B cells. Briney recounted that for many years, B cells have been sorted into 96-well plates using fluorescence-activated cell sorting (FACS), and heavy and light chains have been amplified using single-cell PCR. These procedures, or possibly more recently developed alternatives, allow selected B cells to be studied genetically and functionally. Such work has already led to a clinical trial of an early vaccine candidate.
Looking at B cells—and T cells, too
Briney’s group is just one of many research groups that has started using faster, higher throughput, lower cost platforms for single-cell analysis. Another one of these groups is the Laboratory for Systems and Synthetic Biology (LSSB) at ETH Zurich, where Alexander Yermanos is pursuing a doctoral degree in biosystems science and engineering. When asked about the PepTalk conference, Yermanos described how the LSSB, which is led by Sai Reddy, PhD, first used FACS and later the 10X Genomics platform to query the immune repertoire. The lab uses two different models—B cells from virally infected mice, and tumor-infiltrating lymphocytes—to look for antibodies and T cells, respectively, that are reactive against the virus or tumor.
“We’re keeping the pairing information,” said Yermanos. The LSSB investigators also have transcriptome information from the 10X Genomics’ platform, so they can look at the gene profile of the B or T cells as well as the actual adaptive immune receptor.
“If you expected a T cell to be tumor reactive, for example, you would expect it to have an activation signature phenotype on the transcriptome level,” Yermanos noted. “You can see if the T cells with the same sequence [and derived from a single clone] cluster in the same area.”
“We’re trying to use the transcriptome information to guide us to which T cells are specifically and potently tumor reactive,” he continued. Yermanos and colleagues also hope to use the transcriptome information to guide them to rarer immune cells. These cells, besides representing populations that have expanded less, may still be reactive.
This work would not have even been possible at this scale—which involves the analysis of many thousands of cells over the course of a few days—just a couple of years ago, Yermanos exclaimed.
Coating the walls of miniature wells
“By the time affinity maturation to an antigen is completed, that patient’s B-cell response has generated probably an excess of 106 or even 107 unique B cells,” commented Vu L. Truong, PhD, the founder, CSO, and CEO of Aridis Pharmaceuticals. His company is looking for highly neutralizing antibodies, for example, those involved in generating the rare protective response to cancer or pathogens like the Ebola virus.
It’s easy enough to generate a cDNA library of the entire repertoire and then use phage display to assess which is most neutralizing. But the problem is, you don’t get information such as binding kinetics. “The best way to do this,” said Truong, “is in a massively parallel functional screen at the single-cell level” to look at the binding kinetics in every cell, individually, all in one setting.
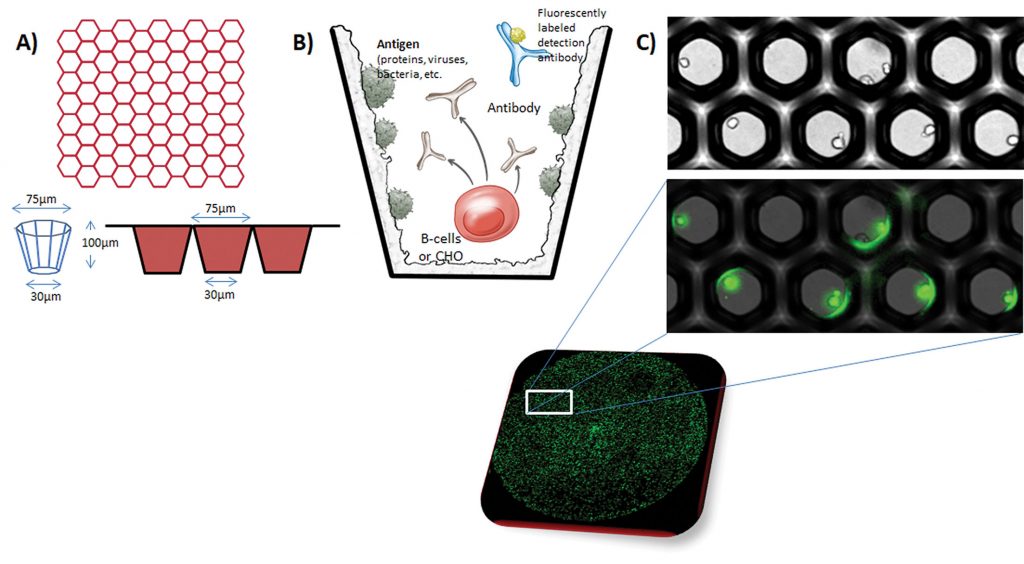
Aridis uses silicon wafer technology to miniaturize the cell culture well—accommodating up to three million wells in a space where there would otherwise be 96. “In the single plate, we can capture every single B cell in the repertoire, [each] in about a half nanoliter volume,” Truong asserted. The technology allows the well walls to be coated with “anything we want,” he continued, and “when you place a B cell in there, within about 12 hours, you can detect the antibody that is secreted from the cell.”
It binds to the well where the antigen is, and it can then be tagged with a labeled secondary antibody. Wells are analyzed by high-content fluorescence imaging to determine which has the highest fluorescence. The cells can then be plucked out with a microneedle, plated singly in microwells, grown, and expanded.
“We have also developed a neutralization assay,” he noted. “Within about two to three days, we can tell you which one of those B cells is most neutralizing.”
Monitoring interactions over time
The xPloration platform, from xCella Biosciences, uses time-lapse images of B cells—up to 1.5 million cells in 40-µm-wide telephone poll–like microcapillary features—to monitor B-cell reactions and interactions over time. This functionality can help investigators find therapeutically relevant antibodies. “We also put in there a target, such as a protein on a bead or—something we’re good at—a cancer cell that has a target of interest,” said Bob Chen, PhD, xCella Biosciences’ director of engineering and co-founder.
“The B cell will secrete an antibody that binds that cancer cell,” he continued, “and then something happens, something simple—it binds properly in the right format, so perhaps it can kill or inhibit the cancer. The target cell can also be a reporter cell. . . . We can look at GFP expression with various reporter pathways. We can look at brightfield findings—morphology, shape, other label-free measurements. Or we can look at cell surface binding using a very simple reaction with fluorescent antibodies.”
This allows for a variety of assays, querying chemical (enzymatic) reactions, cell function, or cell-to-cell interactions. The company is developing activation assays as well.
Once wells of interest are identified, the cells can be instantaneously ejected using a high-throughput laser cavitation process, barcoded, and put into a next-generation sequencing workflow. “All the sequencing we do is based on a single cell that has already passed some sort of metric, like functional status,” Chen pointed out. “We do a screen on cells first, so you’re looking only at the functional variants.”
Doing it all, modularly
The wave of single-cell technology being used for therapeutic antibody discovery generally breaks down into two camps: those that do on-chip genomics, and those that do on-chip phenotypic assays. At the conference, Berkeley Lights unveiled upgrades to its Beacon platform that let it do both.
“We’ll be performing functional screens on-chip [while] also doing a lot of genomics on-chip,” said Anupam Singhal, PhD, the company’s product manager, antibody therapeutics. “We’ll have integration of genomics capabilities that allow you to recover large numbers of sequences once you have identified the relevant hits.”
What’s most important when you’re screening many cells is that you are also recovering the relevant diversity of the population, rather than just the most abundant clones. “Our technology,” he stressed, “will take in the diverse population. It will do relevant deep screening to identify the actual suitable candidates, and we’ll get them all back.”
The Beacon optofluidics chip features thousands of 250-pL reaction wells (called NanoPens) where individual cells can be cultured and assayed. Light is used to deterministically move cells in and out of the pens and into microfluidic channels. Chips can be processed in parallel, or modules can be ganged together serially to assess different functional aspects of a given cell.
Singhal presented an example of using Beacon to search for the rare antibody. “To do that,” he detailed, “you need ways to downselect” using a series of screens, otherwise you end up having to do sequencing, re-expression, cloning, and a lot of manual labor on many more leads. In this case a (secondary) cell-binding assay and a (tertiary) blocking assay are added to the (primary) hit screen. “We went, in a single experiment, from tens of thousands of cells, to 600 hits, to 300 that bind the actual cell target, to 47 that were actual functional lead candidates,” he reported. “That was all done in a single day.”
