
August 1, 2012 (Vol. 32, No. 14)
Real-time quantitative PCR (RT-qPCR or qPCR) is revolutionizing many aspects of molecular biology and becoming the gold standard for accurate, sensitive, and fast quantification of gene expression. Relatively new, it also is experiencing growing pains.
Key challenges right now include finding the right reference gene and improving data analysis. As its popularity grows, new-kid-on-the-block technologies such as two-dimensional RT-qPCR and multiplex digital RT-qPCR are proliferating.
The ability to capture a two-dimensional (2D), spatially accurate expression profile of tissue provides a unique way to follow molecular pathologies. Michael Armani, Ph.D., developed, with his colleague Michael Tangrea, “a 2D-RT-qPCR methodology that quantifies RNA across tissue sections in a single platform.” Dr. Armani is a post-doctoral research assistant, lab of pathology, National Cancer Institute, and was a presenter at CHI’s recent “Quantitative Real Time PCR Conference for Molecular Diagnostics”.
“Overall, the purpose of this approach is the quantification of target mRNAs corresponding to their original positions within the two-dimensional tissue architecture,” he said. The researchers use a 384 well plate over which tissue sections are laid. Then through a sequential series of reactions, tissue is lysed, RNA isolated and reverse-transcribed, followed by qPCR.
“In collaboration with Elisabeth Smela and Benjamin Shapiro at the University of Maryland, we developed a method that uses a grid format in a multiwell plate to macro-dissect tissue sections in order to better preserve the spatial locations of the RNA. We developed and validated the 2D-RT-qPCR in 384-well plates using magnetic recovery beads for RNA isolation in a volume of 20 microliters. Use of such a small volume for tissue lysis and purification had not been done before. The first challenge for us was to characterize the purification efficiency.”
One of the major benefits and applications Dr. Armani envisions for this technology is the ability to mine RNA data in existing tissue banks to better understand pathologies and treatments. “Our end goal is to improve treatment decisions. Usually pathologists are microscopically studying and drawing conclusions on the type, grade, and prognosis for cancers. Although this is a 100-year-old technique, it is still the most actively used and robust method for diagnosis or treatment.
“Staining slides is inexpensive, consistent, and effective. However, considering that there are more than 20,000 genes that exist in humans, blocks of stored tissue essentially have locked up within them an incredible source of information particularly as to potential mutations and genetic patterns.”
The next goals for the project are to ramp up the number of gene-expression patterns. “Initially we provided proof-of-concept with one gene versus control. Then we did three, now we are up to 24 in each well. What’s needed next is to develop even finer resolution. ”
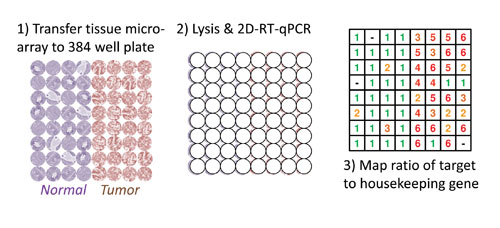
The schematic shows a section from a tissue microarray (TMA) applied to a new technique (2D-RT-qPCR) to study mRNA gene expression across several dozen archival samples. First, the TMA is sectioned onto a tape backing (not shown) and applied to a commercially available 384-well plate. In the second step, lysis and purification is carried out in each well while maintaining the layout of the TMA. Finally, the qPCR detection thresholds for a target gene relative to a housekeeping gene can be displayed as one possible output format for quick analysis of many samples. [National Cancer Institute]
Multiplex Digital RT-PCR
Digital PCR provides a quantitative analysis of nucleic acids as it detects single molecules of DNA or RNA. “Compared to real-time qPCR, digital PCR provides absolute quantification of nucleic acids and potentially performs at a higher accuracy,” says Feng Shen, Ph.D., director of R&D, SlipChip.
“While digital PCR has been demonstrated on a variety of platforms, these platforms usually require complex control systems for sample manipulation and fluidic control. This could make digital PCR less routinely accessible in laboratories or resource-limited settings. The SlipChip is a microfluidic device that consists of two plates with imprinted wells, and the fluidic path can be established or broken apart by simply bringing wells on different plates in or out of contact. SlipChip can generate a large number of reaction compartments, which is required by digital PCR, by relative movement of the two plates, or a ‘slipping’ step.”
Targeting point-of-care and resource-limited settings, Dr. Shen and colleagues developed a multivolume digital RT-PCR SlipChip in Professor Rustem Ismagilov’s laboratory (currently at California Institute of Technology) to assess viral HIV and hepatitis C (HCV) loads. “Although the use of antiretroviral treatments has become more common, it is important to evaluate viral load from patients periodically to prevent the spread of drug resistance,” Dr. Shen explains. This requires quantitative measurements of viral RNA over a large dynamic range. We developed a microfluidic rotational SlipChip to perform multivolume digital RT-PCR with a dynamic range from 1.7×102 to 2.0×107 molecules/milliliter with threefold resolution and lower detection limit of 40 molecules/milliliter.
“Instead of using a large number of wells with uniform volume, we used far fewer wells with different volumes (from 0.2–625 nanoliters) to address the need of a large dynamic range. The tests were conducted using synthetic RNA and validated in a multiplex format by HIV viral RNA and HCV control viral RNA.”
The team found that the results of the viral load test on the SlipChip were self-consistent, and the HIV viral load tests of clinical patients’ samples were in good agreement with the results determined by the Roche COBAS Ampliprep/COBAS TaqMan HIV-1 Test.

The multivolume digital RT-PCR SlipChip is designed for quantification of nucleic acids with a large dynamic range. (Size comparison to a U.S. quarter.) It can be used to quantify multiple analytes simultaneously, or analyze single samples at a dynamic range of 1.7×102 to 2.0×107 molecules/mL with threefold resolution and lower detection limit of 40 molecules/mL.
Which Reference Gene?
Quantitative gene-expression studies performed with RT-qPCR require normalization of target genes using reference genes that are stable and independent of variables tested in experiments. However, there is no one reference gene that is universally applicable to every study, says Virginia Rebecca Falkenberg, Ph.D., microbiologist, Centers for Disease Control.
“Although a number of reference genes have been described for a wide variety of tissues and experimental conditions, they are not universally suitable or necessarily even present in all samples. For example, common reference genes are lacking for RNA derived from whole blood as well as isolated peripheral blood mononuclear cells (PBMC). Further, there are no suitable reference genes for analysis of many chronic and neuropsychiatric diseases for which blood may be the only sample to study gene expression as a systemic sensor of multisystem pathology.”
Dr. Falkenberg and associates initially were working on large-scale clinical studies for biomarker identification for various diseases. “We had received both PBMC and whole blood from patients that were obtained at the same time points. It is very unusual to have access to both. We set out to identify a suitable common reference gene. We first performed a literature review and analyzed our unpublished microarray datasets. We generated a set of six possible reference genes. We then used two algorithms (geNorm and NormFinder) to find the most stable genes for both PBMC and whole blood RNA.”
Next, Dr. Falkenberg changed an additional analysis program in an important and instructive way. “I modified a method called single control normalization error (E). Data is collected for several potential reference genes. To select the best (most stable) genes usually one takes each proposed reference gene and performs statistical analysis comparing one gene at a time to that potential reference gene. I decided to perform analyses by comparing the average of 2–3 genes simultaneously with the proposed reference gene.
“To allow the selection of a reference gene that works in both whole blood and PBMC, I took a single gene or average and the comparison was between the two different tissues instead of two different genes. This greatly improved the process allowing us to identify Phosphoglycerate kinase 1 (PGK1) as one of the optimal normalization genes for both whole blood and PBMC RNA.”
The idea of being able to utilize a single reference gene provides a much more cost-effective means for characterizing gene-expression profiles. “Using a single reference gene also improves the ability to compare gene-expression results using blood RNA collected and processed by different methods with the intention of biomarker discovery.”
Rapid Analysis
Computation is a crucial step to measure gene expression for RT-qPCR. “Data analysis is not straightforward since it requires many iterative computations for data normalization and optimization,” suggests Daijun Ling, Ph.D., scientist, USC Davis School of Gerontology, University of Southern California.
“Oftentimes, PCR machines provide instrumental software that also functions in data analysis. Many just use what the companies provide rather than more suitable software.”
Dr. Ling developed an all-in-one computer program based on statistical software called SAS (SAS Institute). “I called this SASqPCR. It provides multiple macros to assess PCR efficiencies, validating reference genes, optimizing data normalizers, normalizing confounding variations across samples, and statistical comparison of target gene expression in parallel samples.”
SASqPCR does not require the user be an expert programmer. Rather, the program provides a dynamic interface for user-controllable customization based on specific research aims, data quality, and experimental design. Users can easily test a variety of combinations of different analytical scenarios or even customized analytical processes.
“The advantage of the SASqPCR program for analysis of RT-qPCR data is its versatility. First, SASqPCR has no limitation to the size of the dataset. It also can be used for profiling transcriptomes generated by RT-qPCR, as well as population genetic studies with hundreds of subjects.
“Second, SASqPCR can be easily combined with other SAS procedures for advanced statistical analysis, for example, cluster analysis for transcriptome profiling, or association studies for connecting gene-expression variation with particular biomedical conditions. Users are the final decision-makers in analysis of their data.”
Multiple combined datasets or PCR array data can be analyzed. “Users can manage and analyze data in a more traditional way instead of having to rely on proprietary instruments or even plate-based data formats. The final results are basically numeric values that provide a much more straightforward and accurate means to determine how to visualize their results.”
Malaria Diagnostics
Malaria (Plasmodium) continues to remain one of the most devastating infectious diseases on the planet. While new initiatives aim to eradicate the disease, one important aspect of these projects is the necessity for improving diagnosis and surveillance methods. Despite inroads in molecular approaches, the gold standard for malaria diagnosis, epidemiology, and clinical trial efficacy evaluation continues to be microscopy, in particular the Giemsa-stained thick blood smears.
“Microscopy suffers from limitations such as sensitivity, with the average microscopist able to detect about 100 parasites per microliter of blood, but the threshold for fever and clinical disease is less than 10 parasites per microliter for nonimmune patients,” says MAJ Edwin Kamau, Ph.D., chief of the department of molecular diagnostics, Malaria Vaccine Branch at Walter Reed Army Institute of Research.
“As we move into an era working to control and eliminate malaria, it is critical that we develop a more sensitive and higher throughput means for detecting smaller amounts of the parasite useful for even subclinical infections. The malaria vaccine and drug development program at the Walter Reed Army Institute of Research is seeking better means of detection, particularly for those subclinically infected. To do this, we sought to develop a highly sensitive genus-specific RT-qPCR assay to detect Plasmodium.
“A number of such assays have been developed that can measure infection more than 1,000 times more sensitively than microscopy or even antigen detection tests. However, one challenge is that most qPCRs target the DNA of the multicopy 18S rRNA genes, which have enough genetic variation to be problematic. It is therefore important that qPCR target amplification of conserved regions of 18S rRNA genes.”
Dr. Kamau and colleagues developed a genus-specific reverse transcriptase RT-qPCR assay to detect Plasmodium. “The assay detects both rRNA and DNA and takes advantage of the high copy numbers of rRNA in the genome of the parasite. It can detect 0.002 parasites per microliter of blood, one of the lowest levels detected so far.”
According to Dr. Kamau, introduction of the reverse transcriptase step was key. “We were able to improve the assay by approximately 10-fold by adding this step. We verified our hypothesis that RT-qPCR was more reliable than microscopy at lower parasite densities, but not at higher densities. Thus another important finding was that dilution ranges of the sample are important.”
Before molecular diagnostic approaches replace microscopy, there are a number of remaining challenges to overcome including the need for stringent standardization of the methodology.

