
May 15, 2013 (Vol. 33, No. 10)
Post-translational modifications (PTMs) collectively pertain to a phase in protein biosynthesis that involves changes in a polypeptide chain, resulting in a fully functional protein product.
The completion of the Human Genome Project has generated an extensive compilation of genotypic information, yet these sequences further undergo adaptations during translation, producing a wide variety of proteins that play specific roles in the normal physiology of the cell. Current proteomic efforts have thus focused on understanding how cellular activities are governed by proteins.
Proteomics, or global protein analysis, appears to be more challenging than genomic analysis. First, the isolation of intact proteins and the analysis of each amino acid within a polypeptide chain are often plagued by issues relating to its biochemical properties. Advances in instrumentation have identified mass spectrometry as the most promising technology in protein analysis.
Coupling this technique with computational tools has allowed scientists to further enhance the speed and accuracy of protein identification, characterization, and quantification. Also known as “next-generation proteomics,” this approach has been developed to complement the existing next-generation DNA sequencing technology.
The application of mass spec to large-scale PTM profiling of O-GlcNAcylation and phosphorylation proteins in the mitochondria as well as the elucidation of their important roles in the development of diabetes and heart failure has relied on the use of chemoenzymatic labeling for the enrichment of protein samples for better detection and analysis.
“The detection of O-GlcNAc modifications remains a bottleneck that restricts further research in this field,” explains Junfeng Ma, Ph.D., a postdoctoral research fellow at the laboratory of Gerald W. Hart, Ph.D., at the department of biological chemistry, Johns Hopkins University School of Medicine. The detection of O-GlcNAc modification can be done by classical biochemical assays (e.g., O-GlcNAc-specific antibodies) and by mass spec. However, mass spec is the only powerful and high-throughput tool for site mapping.
“The difficulties of unambiguous assignment of O-GlcNAc modification sites in proteins lie in two major aspects: 1) the glycosidic bonds are liable in gas phase and therefore the O-GlcNAc moiety is lost with the traditional collision-induced dissociation (CID) mass spectrometry; and 2) the O-GlcNAc modification is generally of rather low abundance, making it even more challenging to detect,” says Dr. Ma.
Dr. Ma and others tried to address these issues. After successful trials of several methods, they developed a more robust and reliable method that can be applied to diverse samples by more laboratories worldwide. Dr. Ma describes a newly refined method for the enrichment and identification of O-GlcNAc proteins.
For this method, 1) O-GlcNAc groups in proteins/peptides are tagged with azido sugars by using a mutant galactosyltransferase; 2) with the use of a multifunctional reagent, the tagged peptides are captured via the click chemistry on solid phase followed by mild release with chemical cleavage; 3) the released peptides are readily detected by electron transfer dissociation mass spec, which can keep the O-GlcNAc moiety intact and therefore is very useful for O-GlcNAc detection, as demonstrated by collaborator Weihan Wang, Ph.D., at the chemistry department, University of Virginia.
In comparison to previous methods (e.g., antibody-based enrichment and hydrophilic chromatography), the newly developed capture-and-release approach shows higher selectivity, specificity, and sensitivity. The applicability of this method has been tested with individual proteins, mitochondrial samples, and others. Of particular note is that for the first time, they have identified tens of O-GlcNAc proteins in mitochondria.
“By combining the new O-GlcNAc enrichment method and the well-established phosphopeptide enrichment methods, we are doing a larger-scale PTM profiling of O-GlcNAcylated and phosphorylated proteins in mitochondria, which might provide a novel insight for the elucidation of the etiology and development of diabetes and the related diabetic cardiovascular diseases,” Dr. Ma concludes.
Glycosylation
An on-tissue mass spec imaging method has facilitated analysis of glycosylation PTMs associated with different cancers for E. Ellen Jones, Ph.D., a postdoctoral fellow in the laboratory of Richard Drake, Ph.D., professor and director of the Medical University of South Carolina (MUSC) Proteomics Center, and her colleagues.
Through a collaboration with Anand Mehta, Ph.D., of the Drexel Institute for Biotechnology and Virology Research, and Thomas Powers, a graduate student at MUSC, they have extensively characterized the precise glycan structures and sites of glycosylation.
“This new mass spec-based glycan imaging approach combined with on-tissue protein N-glycanase F (PNGaseF) digestion allows us to spatially profile released N-linked glycans in their local microenvironment. The method has been designed to facilitate the tissue analysis of the cell surface glycan changes that occur with cancer progression and other diseases, while maintaining pathology-compatible preparation workflows,” says Dr. Jones.
She further explains that matrix-assisted laser desorption/ionization mass spec imaging (MALDI-MSI) has primarily been utilized to spatially profile proteins, lipids, drug, and small molecule metabolites in tissues, but it has not been previously applied to N-linked glycan analysis.
“Using this basic approach, global snapshots of major cellular N-linked glycoforms are obtained, including their tissue localization and distribution, structure, and relative concentrations. Depending on the tissue type used, generally 20–30 N-glycan species are detected simultaneously.
“The method has been applied to multiple frozen tissues, with emphasis on human prostate cancer, renal cancer, and different therapeutic mouse xenograft cancer tissues. Our initial data suggests that this method can be used to identify multiple N-glycan species reflective of the many changes that occur during the transition of organ-confined tumors to the metastatic phenotype, linked directly to histopathology data,” discusses Dr. Jones.
She also explains that off-tissue extraction of glycans from similarly processed tissues, permethylation, and further mass spectrometry analysis has confirmed these structural designations.
“The glycan profiles are also readily applied to MALDI-MSI imaging data of proteins and lipids in the same tissues, moving toward a more complete biomolecular spatial profile of targeted tissues. These translatable workflows using MALDI-MSI and on-tissue PNGaseF digestions are in continued development, and can readily be applied to any tissue type of interest,” mentions Dr. Jones.
Efforts are also ongoing to use the mass of the identified glycans to identify the specific glycoproteins that were carrying the glycans using high-resolution glycopeptide analysis approaches on a Thermo Scientific Orbitrap Elite mass spectrometer.
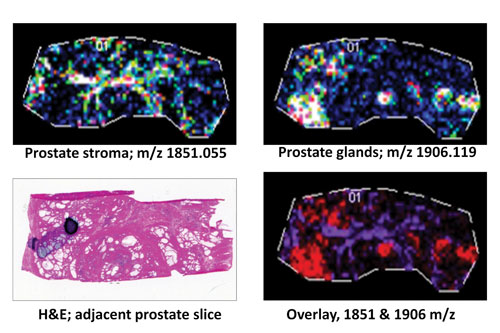
A frozen human prostate tissue slice (10 µm; distal nontumor) was digested with protein N-glycanaseF to release N-glycans: Expression profiles of two representative released N-glycans specific to stroma (m/z 1851.055) or glandular regions (m/z 1906.119) are shown individually or as an overlay in comparison to an H&E stain of an adjacent prostate tissue slice. [MUSC]
Data and Algorithms
In terms of using algorithms and software in investigating PTMs, several scientists have embarked on different approaches in capturing as much information across various tissues and conditions. Nuno Bandeira, Ph.D., assistant professor at the Skaggs School of Pharmacy and Pharmaceutical Sciences and the Department of Computer Science and Engineering at UC San Diego, focuses his research on the premise that various PTMs generate characteristic signatures in mass spectrometry data.
“We want to conduct our investigations using a broad as possible approach and dig deep into any direction that we could go. There is so much variation across different tissues, as well as modifications in peptides, that this gap between biological PTMs and spectral libraries is widening with massive efforts in both realms,” he explains when describing the many advantages of searching mass spec data against spectral libraries (collections of reference spectra) instead of using the more traditional database search approach.
Dr. Bandeira is currently developing an even larger compendium of proteomics mass spec data that will house PTM data from different issues. “This MassIVE (Mass spectrometry Interactive Virtual Environment) community-wide repository we are developing will serve as a resource to converge all existing information on PTMs and mass spec data, including data from the latest types of mass spec instruments. Current databases provide limited information in that generally, these do not offer raw data associated with a mass spec report or vice versa.”
Dr. Bandeira has also expressed his hopes that researchers in the field of proteomics engage in data sharing. “Our vision in setting up this MassIVE repository at UCSD is that researchers will be able to easily access, upload their data, and even search what is available at the site. Even in its current version, our ProteoSAFe system has already enabled the analysis of over 1 billion spectra from over 3,000 users. We are aware of how important spectral data is with regard to PTMs and thus putting all this information together in one site, with users having free access to the information, will definitely enhance the analysis and understanding of PTMs,” says Dr. Bandeira.
For Marshall Bern, Ph.D., vp of Protein Metrics, screening for PTMs can be very challenging because it highly relies on prior knowledge and existing databases. “The current list of reported PTMs is now larger compared to previous years, and this is due to the rapid improvement in analytical tools in proteomics. Although this may be a major advancement in the field, this also increases the need for a reliable and rapid algorithm that understands what peptides one is trying to identify.
“With the knowledge that each amino acid occurs at multiple sites along a polypeptide chain, multiplied by the modification that marks each amino and changes its mass state, then you end up with a larger number of protein possibilities to search in a database. Since PTMs are generally characterized by mass states, if you have a longer peptide, then you will most probably have more mass states per amino acid, thus making it harder to search through a comprehensive protein resource,” says Dr. Bern.
Protein Metrics reports that it offers one of the best solutions in searching databases for protein analysis. Dr. Bern explains that their new proteomics search engine, Byonic, addresses the most common weaknesses in searching databases.
“Byonic offers a wildcard search that assists in finding matches, including specific changes within a peptide such as a PTM on the N-terminus of a chain,” says Dr. Bern. He also explained that although this approach may be slower than known-modification searches, it decreases the chance of missing a PTM by considering every possible mass shift within a range.
“The software also helps in finding matches that carry more than two modifications. Byonic can identify hundreds of times more PTMs than any existing software including N- and O-linked glycans. Byonic lets the user ask and answer more questions since it covers more types of PTMs,” he discusses.
Proteolytic PTMs
Christopher M. Overall, Ph.D., Canada research chair in metalloproteinase proteomics and systems biology at the Centre for Blood Research, University of British Columbia, has investigated proteolytic PTMs extensively.
“Every protein in a cell has been subjected to the activities of proteolytic proteins or proteases, which are highly ubiquitous and comprise the second-largest enzyme family in man. Proteins that undergo cleavage often change their biological function. For example, chemokines, which are chemoattractant cytokines, undergo modifications that change their activity and binding capacity, influencing their role in inflammation and immunity,” says Dr. Overall.
He has also explained that matrix metalloproteinases act on these chemokines, which can render these as an agonist or antagonist to a cell by switching its role as a receptor and enhancing the influx of specific ions into particular cells. In other cases, cleavage of chemokines may even result in its disengagement from its matrix, he adds.
Dr. Overall’s research team was the first to use the yeast two-hybrid screen for protease substrate finding, utilizing inactive mutants of proteases and isolated domains of proteases as bait.
“While effective, it is slow. The field of degradomics, or protease substrate discovery, requires the newest techniques in finding substrates of proteases. Our group has developed highly novel proteomic approaches to specifically identify cleaved proteins in tissues. We are currently funded by the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council to embark on an extensive effort to develop new polymers for proteomics by using high-throughput selection of modified peptides,” says Dr. Overall.
Top-Down Approach
One fresh and interesting strategy for the study of PTMs is the top-down proteomics approach, which involves the screening of whole proteins at the level of their primary structure.
Proteins are highly dynamic molecules that are in many ways more difficult to analyze than the genome, according to Neil L. Kelleher, Ph.D., professor in molecular biosciences, chemistry, and the Feinberg School of Medicine at Northwestern University. Furthermore, use of the classical bottom-up proteomics approach of digesting peptides using proteases and subjecting these fragments to LC/MS does not maximize the information that could be gathered.
“The top-down proteomics approach does not involve protein digestion and thus, identification is mainly derived from fragmentation of the intact polypeptide. This major leap in technology development in proteomics will significantly enrich protein identification and characterization efforts,” Dr. Kelleher notes.
“Capturing proteins in their near-natural form not only increases the information content obtained, but may also prevent further confusion with regard to protein terms such as isoforms, variants, and species,” he explains. Dr. Kelleher and a new Consortium for Top Down Proteomics have proposed the use of the term “proteoform” to describe various proteins products generated from a single gene, including those derived from alternative splicing, genetic variations, and PTMs.
“The many previous terms did not describe cleanly the wide range of protein forms that have been discovered and reported, which created some degree of confusion in protein nomenclature. It is apparent that grouping proteins based on their gene origin allows a more direct and informative approach to protein identification in a gene-specific fashion,” he adds.
Dr. Kelleher has articulated a new “cell-based” version of the Human Proteome Project, which would call for defining proteomes based on specific human cell types.
“The plan is to detect and characterize a billion proteoforms emanating from the ~20,000 genes as expressed and processed in the diverse cell types in the human body. Both bottom-up and top-down approaches can be utilized together to address questions in proteomics and human disease with greater precision,” he emphasizes.
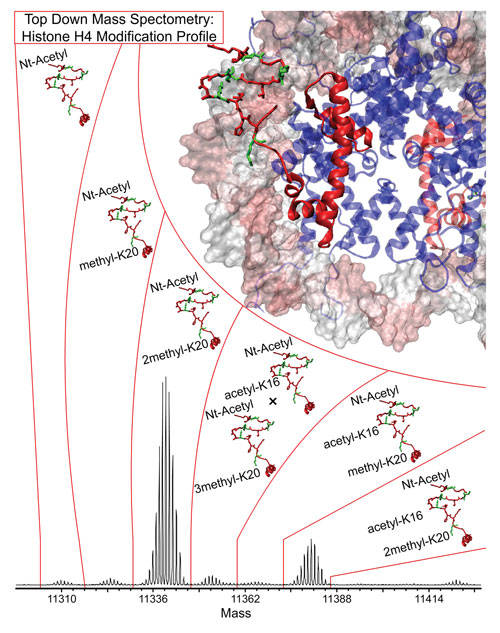
A top-down approach to MS-based proteomics using histone H4: The identification of PTMs in a natural form allows precise identification of proteins without the disruptive effects of protein digestion that is commonly employed in the classical bottom-up approach. [Northwestern University]

