
August 1, 2012 (Vol. 32, No. 14)
In addition to the surprising revelation that our chromosomes harbor fewer genes than originally predicted, the Human Genome Project also unveiled that all individuals share approximately 99.9% of their DNA.
The remaining 0.1% of the genome subsequently enjoyed enhanced scrutiny, as it promised to shed light on interindividual genomic variation, which became one of the most fascinating biomedical topics in recent years.
While single nucleotide polymorphisms (SNPs) were initially the focus of many studies, subsequent discoveries unveiled an additional source of genomic variability, which became known as copy-number variation (CNV). Array comparative genomic hybridization (array CGH) helped characterize copy-number variants and advanced research in several clinically relevant biomedical areas, but the quantitative characterization of copy-number variation remains a technically challenging endeavor.
“We have been using CGH arrays for many years, and they allow the detection of genome-wide copy-number changes, but one thing they cannot detect are copy-neutral aberrations,” says Anniek De Witte, technical marketing manager at Agilent Technologies.
One of the technical shortcomings of array CGH is that it can only measure the total number of allele copies in a sample and, therefore, it is only informative about copy-number changes. Acquired uniparental disomy or copy-neutral loss of heterozygosity, a previously underappreciated modification in which a pair of chromosomes or chromosomal segments is inherited from only one parent, that is increasingly being described in hematologic and solid tumors, would not be detected by this approach.
“The only way to detect these aberrations was by adding SNPs to the arrays, to make the distinction whether two chromosomes are different from each other or not, and this is why we incorporated SNP probes into our CGH arrays,” De Witte explains.
This approach helped investigators unveil significant diversity in several chronic lymphocytic leukemia tumor samples that they examined. In addition, this platform, combined with novel algorithms, is able to determine the fraction of aneuploid cells in cancer samples down to 10% for copy-number changes and down to 20% for copy-neutral changes.
“We wanted to address certain technical questions related to the performance of array CGH on small clinical samples, and the use of ultrahigh-resolution platforms in such samples,” says Mark Basik, M.D., assistant professor in surgery and oncology at McGill University and attending surgical oncologist at the Jewish General Hospital in Montréal.
Dr. Basik and colleagues proposed to test whether the density of array CGH allowed smaller DNA lesions, such as copy-number gains or losses, that could not be seen otherwise, to be visualized.
“The answer is yes,” reveals Dr. Basik. In a recent study that used MCF7 cell lines, investigators in Dr. Basik’s group discovered that the 1M CGH array has a remarkable capacity to unveil micro-copy number alterations, defined as alterations affecting chromosomal regions smaller than 1 Mb.
“More recently, we expanded this approach to clinical samples and found very similar results,” adds Dr. Basik. At the same time, Dr. Basik and colleagues visualized many intragenic breaks and, subsequently, by comparing the data with results from the original study that sequenced the same cell line, they identified approximately half of the gene fusion events that had been defined in that study. “Array CGH could become a valuable screening tool to look for gene fusion events,” reveals Dr. Basik.
An additional question that Dr. Basik and colleagues addressed revolves around the use of whole-genome amplification prior to performing high-resolution arrays. With clinical research increasingly relying on biopsies, which generally provide a very limited amount of biological material, whole-genome amplification is often required prior to the analysis of chromosomal variants.
A comparison between the 244K and the 1M CGH Agilent array platforms revealed that whole-genome amplification introduces many amplification artifacts, such as small deletions, that are much more detectable with the 1M platform than with the 244K platform. “This was surprising, and suggested that the use of whole-genome amplification with very high-resolution platforms is problematic and, in fact, algorithms used to automatically detect aberrations will pick up many artifacts and classify them as real aberrations,” cautions Dr. Basik.

The Agilent Human Genome CGH Microarray is a high-resolution tool for genome-wide DNA copy number variation profiling without amplification or complexity reduction.
Reproductive Advances
An area that has been increasingly transformed by array CGH advances is assisted reproductive technologies. At this time, 50% of the embryos generated by in vitro fertilization harbor chromosomal abnormalities. These embryos usually either fail to implant or, in the few instances when they do, the pregnancy would result in miscarriages or in various disorders.
“Our policy, as a company, is that if one can avoid the conception or the transfer of abnormal embryos, this offers a much better choice for the patient,” says Santiago Munné, Ph.D., founder and director of Reprogenetics. The emphasis at Reprogenetics is on testing couples before they conceive and, if they are carriers, perform pre-implantation diagnosis. “This is followed by the transfer of normal embryos and results in an increased implantation rate,” explains Dr. Munné.
While karyotyping may detect abnormalities that are 10–15 Mb in size, array CGH can achieve a much better average resolution, as low as 1 Mb. Recently, Dr. Munné and colleagues reported that array CGH presents several advantages over FISH and SNP arrays in diagnosing translocations in pre-implantation human embryos, as it allowed the simultaneous survey of all the chromosomes, without requiring genetic testing to be conducted beforehand.
In addition, array CGH allows data to be generated within approximately 12 hours for every genomic region that one wants to inquire. “At this time, there is a consensus that prenatal diagnosis should be performed by array CGH, this is the direction everyone is moving toward, and karyotyping will eventually be phased out,” explains Dr. Munné.
High-Density Probes Perform Best
“Mapping structural variants is not trivial, as there are many complications, and CGH arrays represent a terrific tool to validate CNVs that are detected by other means,” says Michael P. Snyder, Ph.D., professor and chair of genetics at Stanford University and director of the Stanford Center for Genomics and Personalized Medicine. Recently, Dr. Snyder and colleagues conducted a comparative study of different CGH array platforms available commercially.
This work, which examined 12 leading genome-wide copy-number variation detection platforms, including Roche NimbleGen, Agilent, Illumina, and Affymetrix arrays, revealed that they all performed reasonably well and were able to detect aberrations of a few kilobases in size. It also showed that none of these arrays are perfect, and each presents certain limitations, in terms of a gold standard dataset that was used in the study.
“Most chromosomal aberrations 30 kb or larger were detected, but smaller ones are often detected by one platform and may be missed by another,” says Dr. Snyder. “But overall, the arrays containing higher-density probes in known copy-number variation regions fared better, and this is one of the important lessons that emerged,” continues Dr. Snyder.
Clinical Applications
Several articles from Dr. Snyder’s lab illustrate the strength that CGH arrays provide in clinical medicine. By using CGH arrays to map segmental trisomies in patients with Down syndrome, the investigators recently generated the highest phenotypic resolution map for this condition to date. This helped narrow down the chromosomal region associated with Down Syndrome-specific congenital heart disease, a major phenotype that accompanies this condition, to a
Moreover, the phenotypic map helped test specific hypotheses that were advanced in the past regarding the etiology of this condition. “Next-generation sequencing is clearly stepping into this area, but arrays are still the perfect tool for confirmation, and they are cheaper, and orthogonal,” emphasizes Dr. Snyder.
Another recent advancement in Dr. Snyder’s lab was the development of a personalized -omics profile, in a strategy that involved high-accuracy sequencing of several genomes, including his own, combined with the analysis of -omics components, and the examination of personal RNA and protein variants, in a study that included over 3 billion molecular components across multiple time points that spanned 14 months.
“The data predicted that I will develop diabetes, and I did,” says Dr. Snyder, and the early discovery of this condition helped him implement timely nonpharmacological interventions, including dietary changes and increased exercise, which most likely would not have been able to control the condition by themselves had it been discovered later on at a routine checkup.
Reimbursement Woes
Microarray analysis has shown substantial benefits in diagnosing certain congenital disorders. However, one of the challenges with using chromosomal microarray analyses for clinical applications is the inconsistent reimbursement for testing, as third-party payers often consider them to be an experimental tool because of a lack of evidence that microarray testing affects patient management or clinical outcomes.
“There is not a lot of information out there published on this, and this was the reason why we tried to gather evidence to show that microarray analysis actually provides useful information for patients,” says Jay W. Ellison, M.D., Ph.D., medical director at Signature Genomics at PerkinElmer.
Dr. Ellison and colleagues selected specific medical conditions that can be diagnosed with microarray technologies and have specific clinical features that require medical follow-up. Many times, patients diagnosed with these conditions might not be aware of their risk to develop certain medical problems.
“We tallied the frequency with which these actionable diagnoses were made, and it was quite high. Up to one-third of the diagnoses that were made had features that would require specific medical or clinical actions. This was the major take-home message from this study,” explains Dr. Ellison.
In addition, the authors tracked a subset of patients to see whether physicians responded to the test results with specific clinical actions that are related to the diagnosis, and reported that specific and appropriate clinical actions were taken approximately 90% of the time, an aspect that awaits further work to be explored in more detail.
“The best future studies will be within provider systems where one can capture those individuals who were tested by chromosomal microarrays and look at the clinical outcomes that result from receiving the result of the microarray,” explains Marc S. Williams, M.D., director of the Genomic Medicine Institute at Geisinger Health System.
In the short time that elapsed since the extent of genomic copy-number variation was discovered, its role in biomedical areas as diverse as physiological development, disease pathogenesis, pharmacogenomics, and evolution has become increasingly apparent. In parallel, technological advances have fundamentally contributed to measuring specific copy-number variants and characterizing their significance.
These strides promise to catalyze the incorporation of this relatively newly discovered source of interindividual genomic variability into routine clinical applications, forecasting one of the most exciting times that medicine and biology have seen.
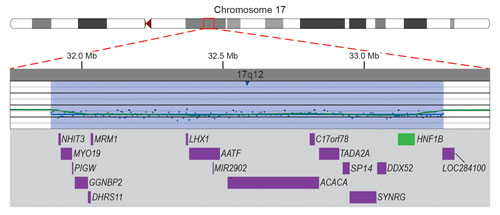
Microarray data demonstrating a submicroscopic deletion of chromosome 17 in a 4-year-old boy with developmental delay. One or more of the genes (purple) in the deleted region (blue-shaded) is suspected to be responsible for the boy’s developmental delay. Deletion of the HNF1B gene (green) puts the boy at high risk of developing renal abnormalities and diabetes mellitus, neither of which was suspected.

