
Wendy A. Lea Ph.D. NIH
In this literature review, the authors show how accumulation of amyloid beta peptides leads to Golgi fragmentation.
First described in 1906 by Alois Alzheimer, Alzheimer's disease (AD) represents the most common form of dementia today and the sixth leading cause of death in the United States (Alzheimer's Association). Symptomatically, this fatal neurodegenerative disorder is most often characterized by loss of memory and decreased abilities in orientation and reasoning. Pathological features of AD include the presence of neurofibrillary tangles (NFTs) and plaques that are formed as a result of accumulation of β-amyloid peptide (Aβ; a proteolytic product of amyloid precursor protein [APP] by secretases). More than 20 million people worldwide currently suffer from this incurable disease, with more than 5 million in the United States. Various factors contributing to AD pathogenesis are being explored, but mechanisms leading to AD remain elusive.
From a drug discovery point of view, several potential targets within the amyloid pathways have been proposed for AD treatment. For example, Fyn, a Src family kinase, is thought to be involved in AD by its manipulated expression level in a transgenic mouse model of AD (Yang et al., J Alzheimers Dis 2011;27:243–252). Association of Fyn to Aβ is also supported by protection against Aβ-mediated toxicity observed in mature hippocampal cells from Fyn−/− mice. On the other hand, it is also thought that Aβ, particularly the soluble Aβ oligomers, are important pathogenic structures in AD, and thus, targeting soluble Aβ oligomers is considered an optimal intervention approach. In this aspect, drug discovery efforts have been placed on generation of antibodies that bind to Aβ oligomers with high specificity and affinity, modulators that efficiently block oligomer assembly, and selective antagonists for neuronal receptors (Hefti et al., Trends Pharmacol Sci 2013;34(5):261–266).
Joshi and co-workers* show in this research article data suggesting that Golgi is a potential drug target for AD. In this study, APPswe/PS1ΔE9, a widely used AD transgenic mouse model was utilized, expressing an APP Swedish mutation and the exon 9 deletion mutant of human PS1. Initially, it was demonstrated that Golgi fragmentation occurred in this transgenic mouse model, as well as in tissue culture cells that were doubly transfected with cDNAs encoding both mutants. Application of specific protease inhibitors to AD cells led to a pronounced reduction in Golgi fragmentation, strongly illustrating Aβ accumulation as a cause for Golgi fragmentation. What could be the mechanisms for Aβ-mediated Golgi fragmentation?
As kinase (cyclin-dependent kinase-5, cdk5) activation has been previously reported as a result of Aβ production, Golgi fragmentation was subsequently reduced in the current study upon the addition of kinase inhibitors to AD cells (Figure). The highly organized stacked Golgi structure is supported by several Golgi structural proteins, such as GRASP65, an important component tethering the cisternae into stacks and a major mitotic kinase target.
The authors further found that GRASP65 was highly phosphorylated in AD cells through immunoprecipitation and Western blot studies, and GRASP65 phosphorylation was due to cdk5 that was activated by Aβ accumulation. After identification and confirmation of Golgi fragmentation mechanism, nonphosphorylatable mutants of GRASP proteins were transfected in AD cells, and Golgi structure and function were rescued. This proof-of-principle study points to molecular causes for Golgi fragmentation and also a potential new target for AD treatment.
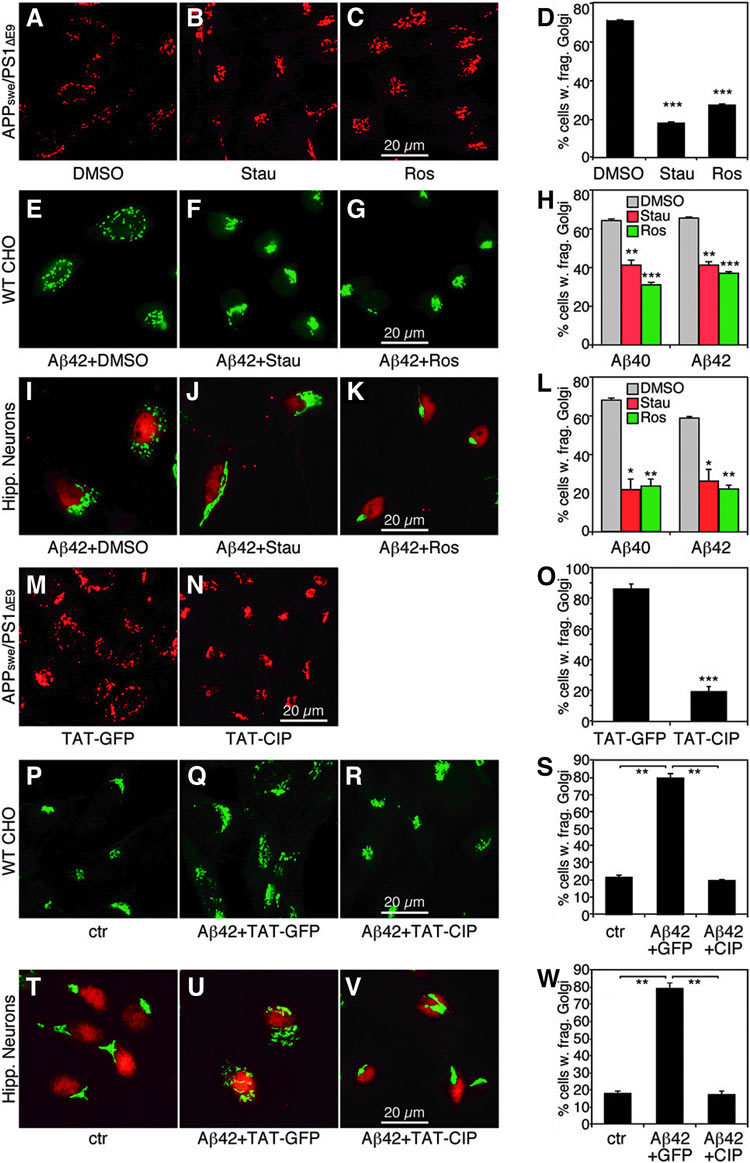
Figure. Kinase inhibitors reduce Aβ-induced Golgi fragmentation. (A–D) Kinase inhibitors reverse Golgi fragmentation in APPswe/PS1ΔE9-expressing (AD) cells. Cells cultured overnight were treated with DMSO (A), 1 µM staurosporine (B), or 15 µM roscovitine (C) for 2 h. Chemicals were added directly to the tissue culture dishes without changing the media. Cells were immunostained for GRASP65, then quantified (D). (E–H) Kinase inhibitors reverse Golgi fragmentation in Aβ-treated WT CHO cells. WT CHO cells were treated with Aβ in the presence of DMSO (E), 1 µM staurosporine (F), or 8 µM roscovitine (G) for 2 h, then stained for GRASP65 and quantified (H). (I–L) Kinase inhibitors reverse Aβ-induced Golgi fragmentation in hippocampal neurons. Primary hippocampal neurons were treated with Aβ peptides in the presence of DMSO (I), 1 µM staurosporine (J), or 8 µM roscovitine (K) for 2 h, then stained for GRASP65 and quantified (L). (M–O) TAT-CIP treatment reverses Golgi fragmentation in AD cells. Cells cultured overnight were treated with 600 nM TAT-GFP (M) or TAT-CIP (N) (added directly into the medium) for 12 h, then immunostained for GRASP65 and quantified (O). (P–S) TAT-CIP inhibits Golgi fragmentation in Aβ-treated WT CHO cells. WT CHO cells were treated with 1 µM control peptide (P) or Aβ42 for 12 h in the presence of 600 nM TAT-GFP (Q) or TAT-CIP (R) added 30 min before Aβ42, then immunostained for GRASP65 and quantified (S). (T–W) TAT-CIP reverses Aβ-induced Golgi fragmentation in hippocampal neurons. Primary hippocampal neurons were treated with either control peptide (T) or 1 µM Aβ42 for 12 h in the presence of 600 nM TAT-GFP (U) or TAT-CIP (V) added 30 min before Aβ42, stained for GRASP65, and quantified (W). *p < 0.05; **p < 0.01; ***p < 0.001, Student t test. DMSO, dimethyl sulfoxide; WT, wild-type; CHO, Chinese hamster ovary; TAT, Trans-Activator of Transcription; CIP, cdk5-specific inhibitor peptide; GFP, green fluorescent protein.
*Abstract from PNAS USA 2014, Vol. 111: E1230–E1239
Golgi fragmentation occurs in neurons of patients with Alzheimer's disease (AD), but the underlying molecular mechanism causing the defects and the subsequent effects on disease development remain unknown. In this study, we examined the Golgi structure in APPswe/PS1ΔE9 transgenic mouse and tissue culture models. Our results show that accumulation of amyloid beta peptides (Aβ) leads to Golgi fragmentation.
Further biochemistry and cell biology studies revealed that Golgi fragmentation in AD is caused by phosphorylation of Golgi structural proteins, such as GRASP65, which is induced by Aβ-triggered cyclin-dependent kinase-5 activation. Significantly, both inhibition of cyclin-dependent kinase-5 and expression of nonphosphorylatable GRASP65 mutants rescued the Golgi structure and reduced Aβ secretion by elevating α-cleavage of the amyloid precursor protein.
Our study demonstrates a molecular mechanism for Golgi fragmentation and its effects on amyloid precursor protein trafficking and processing in AD, suggesting Golgi as a potential drug target for AD treatment.
Wendy A. Lea, Ph.D., works at the NIH.
ASSAY & Drug Development Technologies, published by Mary Ann Liebert, Inc., offers a unique combination of original research and reports on the techniques and tools being used in cutting-edge drug development. The journal includes a "Literature Search and Review" column that identifies published papers of note and discusses their importance. GEN presents here one article that was analyzed in the "Literature Search and Review" column, a paper published in Proceedings of the National Academy of Sciences USA titled "Aβ-induced Golgi fragmentation in Alzheimer's disease enhances Aβ production." Authors of the paper are Joshi G, Chi Y, Huang Z, and Wang Y.

