
April 1, 2014 (Vol. 34, No. 7)
Increasing R&D efficiency is the pharmaceutical industry’s largest challenge as the industry grapples with the major issue of drug attrition.
Killing compounds that should be killed earlier would provide significant cost reductions and improve drug development economics. Recent advances in ADME-Tox assays may be the key.
According to Mark Seymour, Ph.D., the director of science and technology at Xceleron, the inherent characteristics of liquid chromatography and accelerator mass spectrometry (LC+AMS), combined with the use of tracers and innovative clinical designs, can provide an earlier understanding of the kinetics of novel assets.
Ultrasensitive AMS is independent of chemical class, not susceptible to matrix effects (due to the sample-processing procedure), and has a predictable and reproducible lower limit of quantification (LLOQ) in the fg–pg/mL range.
The use of a tracer allows for the generation of fully quantitative metabolism data without the need for reference standards or complicated matrix-matching methods. Concomitant administration of a 14C-labeled intravenous dose with a nonlabeled extravascular dose provides a cost-effective way to generate fundamental pharmacokinetic data, including absolute bioavailability.
AMS is used in all phases of drug discovery and development. For example, early on, AMS can be employed to determine the intrinsic turnover of protein targets to understand their druggability, or for bioanalysis of the cytotoxic “warhead” of antibody-drug conjugates (ADCs).
In Phase 0 human microdosing studies, the technology is deployed to answer specific development questions, such as determining whether preclinical data are equivocal or whether accelerating development of a follow-up compound is required to overcome a lead compound’s specific weakness.
Most Phase I–III applications are conducted at pharmacologically relevant doses during clinical development and include AMS studies in vulnerable populations. Some of these applications are conducted for compounds that are excreted over many weeks, as well as for early assessment of metabolites in safety testing (MIST) liability.
“The largest growth area is in enriched Phase I studies. Often companies first use AMS to generate absolute bioavailability data late in development to meet the requirements or specific requests of regulators. However, once they realize how powerful the data generated can be, there is an increasing tendency to use the approach earlier to provide fundamental information to better direct development,” states Dr. Seymour.

Xceleron’s 250 kV Single Stage Accelerator Mass Spectrometer (SSAMS) is a robust and ultrasensitive bioanalytical tool. AMS technology, originally developed for radiocarbon dating of archeological artifacts, can provide a compound- and matrix-independent platform that enables innovative clinical study designs.
Standardizing Cardiac Liability Data
Drug impacts on cardiac biology—the major reason for drug attrition—can be multifaceted, affecting electrophysiology, energy generation, or function. Current nonstandardized methods use different equipment, skills, protocols, and temperature ranges, plaguing data comparison between laboratories.
“The use of multielectrode arrays (MEAs) to analyze cardiac biology is increasing, driven by the availability of human stem-cell-derived models,” declares Nick Thomas, Ph.D., a principal scientist at GE Healthcare Life Sciences. These models include GE Healthcare’s Cytiva cardiomyocytes and higher-throughput systems. “Human stem-cell-derived cardiomyocytes provide an integrated system to study drug impacts on multiple cardiac ion channels, not just single channels analyzed in isolation,” adds Dr. Thomas, “and MEA is ideally matched to interrogate drug effects in stem-cell models.”
The MEA waveform can be analyzed to extract multiple parameters to provide a phenotypic signature of a drug’s effects, which can be analyzed using hierarchical clustering techniques to rank compounds by mechanism of action and potential clinical risk.
MEA is amenable to standardization, has a higher throughput than traditional manual patch clamping, and has lower skill requirements. Data output is analogous to an electrocardiogram (ECG). MEA-measured field potential duration (FPD) is the equivalent of the QT interval in vivo. If a drug increases the FDP in an MEA, there is a strong possibility that it will prolong QT in a patient.
Cytiva cardiomyocytes can also be used on other analytical platforms, such as imaging systems, allowing analysis of more aspects of a drug’s toxicity on a cell.
“One of the great advantages is that you have complementary platforms, MEA and high-content imaging, both of which produce multiparameter data from the same stem-cell-derived cardiomyocyte model,” asserts Dr. Thomas. “You are more likely to be clinically predictive if you can combine data to give a full signature that encompasses all of the drugs impact on the structure and function of the cell.”
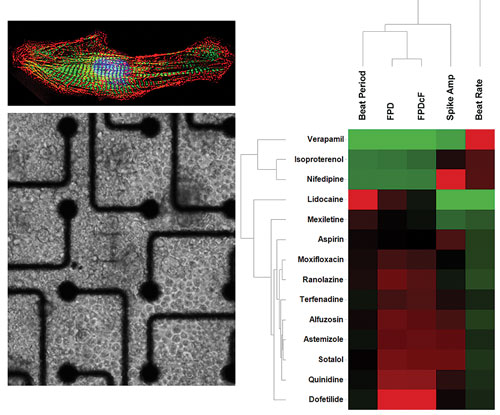
GE Healthcare Cytiva cardiomyocytes showing staining of contractile proteins (top left) and cells growing on multi-electrode arrays (bottom left). An analysis of MEA data ranks clinical drugs for risk of QT prolongation (right).
Increasing Use of HCA
Although in vitro cytotoxicity assays have been around for a long time, only when high-content analysis (HCA) for cell-based assays was introduced were they able to provide good predictions. Today, HCA is used extensively in predictive toxicity for small molecules.
“HCA gives you the ability to study the functionality of live cells. Not many other technologies allow you to do that,” says Peter O’Brien, DVM, Ph.D., DVSc, a lecturer at University College Dublin. “For about 80% of small molecule drugs, HCA is predictive of human toxicities—compared to animal studies, which may turn up only 50%.”
“Pharmacokinetic measurements migrated from animal models to cell-based assays about 10 years ago. As a consequence, attrition due to bioavailability was reduced by an order of magnitude. The same is going to happen with drug toxicity.”
If toxicity affects the fundamental processes of life, then the system’s sophistication is irrelevant. Obvious toxicities are easily detected. Of greater interest is the detection of nonobvious toxicities that affect a small proportion of people.
As HCA becomes more engrained into the routine safety strategy and assessment of candidate compounds for preclinical and clinical development, multitiered approaches will screen for potential toxicities, enhance understanding of the underlying mechanisms, and then rank compounds within a series. In addition, in vivo toxicity can now be measured using HCA.
“We take blood cells from treated animals and then assess these blood cells in the same way as it is done in vitro in drug discovery. Translational biomarkers also are a key future application for HCA. When you have a drug that works at the fundamental level— for example, inhibition of mitochondrial replication—you can detect the biomarker in individual cells, you can monitor it in animals, and you can monitor it in people,” concludes Dr. O’Brien.

Doxorubicin (Dox), an anthracycline anticancer drug, is widely used against a variety of human tumors, but there are associated toxicity risks. Researchers at University College Dublin have demonstrated that high-content analysis of blood cells can be used to identify human toxicity: blood cells that do not exhibit toxicity (left); blood cells that exhibit a toxic effect due to Dox (right).
In Vitro Immunotoxicity Assays
If an immune suppression response is expected, regulatory authorities require immunotoxicity studies for small molecules.
In 1966, the Mishell-Dutton (MD) assay was originally described. Dissociated spleen cells from nonimmunized mice were stimulated in vitro to generate an antibody response similar to that which occurs with in vivo immunization. MD cultures are the in vitro equivalent to the widely used, ex vivo primary T-cell-dependent antibody responses (TDAR), which regulators have identified as a main functional test for immunotoxicological investigations.
The MD assay was first used with mice cells. Recently, it has been adapted for use with rats, dogs, human, and nonhuman primate cells. Cells are cultivated in vitro with the antigen for about one week, eliminating in vivo immunization. The antigen, sheep red blood cells (sRBCs), allows the use of different cell types, such as spleen cells or peripheral blood mononuclear cells (PBMCs).
Incubating the immunized cells with the test substance allows assessment of whether or not immunosuppression occurs through the use of a plaque-forming assay. Currently, the adapted MD assay is used as an additional screening tool. The assay is fast, less labor-intensive, and more cost-effective than TDAR, and allows monitoring of immune state changes over time.
“The TDAR is only performed on mice or rats. You can conclude a lot from rodent species, but to be really sure, it is better to use a human model,” notes Anna Fischer-Berenbein, Ph.D., a postdoctoral fellow at Bayer Healthcare.
“Our initial experiments show that in vitro MD cultures are equivalent to in vivo assays,” observes Dr. Bernbein. “Data are still limited, but as testing continues, we will be able to determine if the assay is truly predictive. If you have the same results in vivo in animals as in vitro in human cells, then you can be quite sure you will have the same results in vivo in humans.”
Mechanistic Models for DILI
DILIsym® software, a modular, deterministic mechanistic model, was specially designed for predictive indications of drug-induced liver injury (DILI).
The foundational components of the model were incorporated from concepts developed in the literature, as well as from basic biological and physiological principals. Since species-response difference is a main driver for model development, underlying biological differences across species—mouse, rat, dog, and human—were built into the equations and parameters.
“DILIsym gives a quantitative prediction of the pharmacokinetics and the drug exposure to the liver over time while representing the basic biological liver processes known to be involved in DILI. Bringing these factors together, and mathematically integrating them into a common platform, makes the application powerful,” explains Brett Howell, Ph.D., the lead scientist and manager of the DILI-sim Initiative at the Hamner Institutes for Health Sciences.
Many biological processes are nonlinear. Some reactions happen quickly, and others, such as liver regeneration, can take up to a year. Although managing these timescales requires more computational power, DILIsym can be run on a desktop computer.
The user inputs the dose, species, and compound-specific parameters that can be obtained with in vitro experimental systems. Then the software models how the liver is exposed to the drug over time. The software, which can accommodate either single-dose or multidose protocols, can predict how the liver will react (including the death and regeneration of cells) as well as the behavior of clinical biomarkers.
In general, the model has utility in all phases of drug discovery and development. Users can tease out mechanistic reasons for getting species-specific responses and, ultimately, determine which response is more human-like for a particular compound.
“The economics tell us we have to become more efficient in developing drugs. Killing compounds that should be killed earlier would be the biggest source of cost savings. But there are safe drugs that are killed that we need to let through. If there is a signal in a rat that has nothing to do with human biology, then we should not be killing that drug,” concludes Dr. Howell.
Carcinogenicity Studies
One of the main drags on ADME/TOX research is that carcinogenicity studies can run as long as two years. Why is this the case?
“The tumorigenic potential and possible human hazard of pharmaceuticals with expected continuous or repeated use or with cause for concern needs to be assessed before entering large-scale clinical trials,” says Nico Scheer, Ph.D., head of the tADMET™ portfolio at Taconic.
The traditional approach for such carcinogenicity studies is the 2-year bioassay in rats and mice, he explains, adding that these lifetime assays have several limitations, such as the protracted study duration, the large number of animals required, the high costs, the high morbidity and mortality rates, the high number of false positive results due to spontaneous tumor incidences, and the often questionable relevance to human risk assessment.
To overcome these limitations researchers in industry and regulatory authorities have looked for alternatives to the 2-year rodent carcinogenicity assay for a long time. This led to the development and extensive validation of different transgenic mouse models, namely rasH2, p53+/- and Tg.AC. These models were finally accepted by the FDA and other regulators as alternatives to the 2-year mouse carcinogenicity study and are distributed by Taconic, notes Dr. Scheer.
“While the use of Tg.AC is no longer recommended and p53+/- is used only rarely, rasH2 is often the model of choice and research indicates that more than 50% of all mouse carcinogenicity protocols submitted to the FDA in 2013 were carried out in rasH2,” he says.

