March 15, 2013 (Vol. 33, No. 6)
Advances in genomics technology have made sequence analysis of diverse organisms routine. Scientists armed with whole genome sequence data can now ask questions about genome structure and organization that impact gene expression.
Improved access to bioinformatics tools enables scientists to process, manage, and store raw data, annotate sequences to define genes, and quantitate RNA sequence data to get insight into gene expression patterns.
The progress being made by a number of companies and academic researchers enabling resolution of questions from genomic analysis was shared at the recent “Plant and Animal Conference” in San Diego.
Amy Radunz, a nutritionist from the department of animal sciences at the University of Wisconsin in River Falls, has partnered with molecular biologist Hasan Khatib, an associate professor at University of Wisconsin-Madison, to ask how differences in maternal diet during pregnancy induce gene expression and DNA methylation changes in fetal tissues in sheep.
The experiment called for the use of 15 ewes that were fed three different diets during gestation at the same energy intake. The control group was given haylage; this high fiber diet is the most common food source for sheep. The second group was given corn, which provides a high starch diet, and a third group was given distillers product, a byproduct of the ethanol industry, which provides a high protein, high fiber, and high fat diet. Each ewe was kept in a separate pen under standard temperature and exposure to light to control conditions including food intake and the influence of exercise on the transfer of nutrients to the growing fetus.
On day 130 of pregnancy, nonsurvival surgery was performed on the ewes to enable the measure of uterine blood flow to monitor nutrient supply to the fetus and to collect tissue samples from four fetal tissues. Unlike previous studies in cattle and sheep, the different treatments did not confer a noticeable difference in fetal weight.
The working hypothesis going into the experiment was that the primary impact on the fetal growth in previous studies was the result of the amount of glucose in the maternal diet. The high starch diet (corn) provides the greatest amount of glucose, whereas the distillers product provides both high protein and high fat, both of which stimulated greater maternal insulin production in the circulation but did not result in differences in maternal glucose circulation. The conclusion from this study where maternal nutrient supply was measured is that glucose supply to the fetus was not impacted by maternal glucose intake.
The current view is that the relative amount of different amino acids (arginine, methionine, and cysteine) may play a more significant role given that these amino acids contain methyl donor groups that can impact the activity of maternally and paternally imprinted genes.
The impact of maternally expressed genes was looked at for H19, IGF2R, GRB10, and MEG8. The study also looked at the impact of paternally expressed genes: IGF2, PEG1, PEG3, DLK1, and DIO3 for transcriptomic and epigenomic alterations of the imprinted genes in fetal tissues.
Glioma Tumors
Dinorah Friedmann-Morvinski, a post-doctoral fellow in Inder Verma’s lab at the Salk Institute, has shown how tumors originated from mature neurons dedifferentiate into glioma stem-like progenitor cells. This reprogramming process has also been reported in intestinal cancer cells.
Using a lentiviral vector containing either H-Ras oncogene and a hairpin targeting p53 or both tumor suppressor genes, NF-1 and p53, Dr. Friedmann-Morvinski can induce the formation of a glioma tumor in mice injected in the brain. However, since H-Ras is hardly mutated in human gliomas, this oncogene only serves as a surrogate for EGFR, which is overexpressed in gliomas (EGFR amplification leads to Ras activation, hence Ras is a good surrogate for EGFR).
The induction of the dedifferentiation occurs only when two hits occur in the same cell: either H-Ras overexpression and loss of p53 or loss of tumor suppressors NF1 and p53. The loss of NF-1 is relevant to the development of the disease in human patients. NF1 inhibits Ras, hence the loss of NF1 results in overexpression of Ras.
The tumor phenotype in mice takes eight weeks to develop from the H-Ras and shp53 model. However, the loss of NF1 and p53 takes up to seven months to develop the tumor phenotype. The tumor phenotype is revealed when mice appear lethargic and have an enlarged head. Upon histologic examination, GFP markers encoded in the lentiviral vector show up along with disease tissue that reveals GBM, grade 4. These tumors also show stem cell markers, consistent with dedifferentiation.
The tumor cells can be established in tissue culture to form cancer stem cell lines when grown in serum-free media. These cells also have the capacity to form neurospheres, and differentiate into all lineages of the brain (neurons, astrocytes, and oligodendrocytes). As a further indication that these cells have dedifferentiated and behave as cancer stem cells, when transplanted, as few as 10–100 cells injected into mice will form the same glioma tumor type as the primary tumor from which they were isolated.
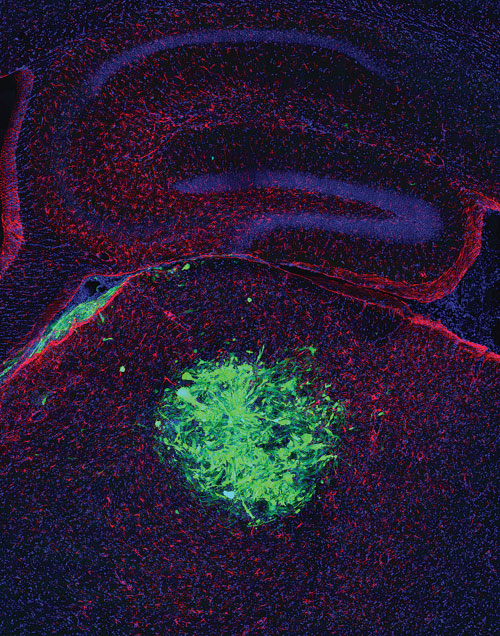
Immunolabeling for GFAP expression marks tumors formed in mouse brains following injection of lentiviral vector in the cortex. [Salk Institute]
Software Tools
At the meeting, Sheldon McKay, iPlant education-outreach-training manager, described the iPlant Collaborative’s effort to develop a cyberinfrastructure platform for biologists.
iPlant infrastructure has been generalized to support life science research; it is agnostic with respect to the species under investigation. The cyberinfrastructure platform is intended to solve basic biological challenges, such as how to analyze complex genomic data and how to analyze gene expression data.
To demonstrate the power of the iPlant Collaborative, researchers are testing a biological model to explain the basis for the heterosis or hybrid vigor that is observed in all crop species and explore the connection of heterosis to cellular aging. This model postulates that hybrids are more vigorous because they possess a greater allelic diversity than their inbred parents and can use this allelic diversity to reduce wasted metabolic energy.
Consequently hybrids are better able to withstand “new” assaults or stresses they may confront, like disease, drought, heat, or salinity. Hybrids have the improved ability to utilize variation in the proteins that affords them protection from the stresses that cause macromolecular damage.
The observation of increased yields, faster growth, and greater longevity seen with hybrids is based on the regulation of energy metabolism and protein quality control. The working hypothesis for this is the reduction of protein turnover, by expressing alleles encoding the most stable proteins, protein degradation is reduced, and energy used for protein metabolism is conserved. The molecular model is that cells with greater than a single allele for each gene express the allele that encodes the more stable protein or efficiently folded protein preferentially.
In the work presented at the conference, two inbred strains of mice (Balb/c and C57Bl) were crossed to look at gene expression patterns in the hybrid offspring at two ages vs. the inbred parents at a young age. Tissue samples from the brain, kidney, liver, and muscle tissue were collected from four animals from each of the parents and the hybrid offspring (young and old).
Data from 64 sets of RNA sequencing runs with 35 to 55M reads per run was collected and analyzed against the mouse reference genome and the transcriptomes of each tissue were compared. The results suggest that older hybrids have a decayed protein quality control system relative to the young hybrids and therefore look more like the inbred parental lines. Given the capability of the iPlant cyberinfrastructure, this complex analysis of numerous large sequence files could be done as a web-based analysis in weeks to months.
The iPlant Discovery Environment features a large number of analysis tools that are integrated by users or the core team.
Through several poster presentations, Genedata showed relevant examples of how the Genedata Selector™ solution, which is a genome data management and analysis system, can be used to handle plant genomic data. The focus is on managing genome and strain information in a relational database.
Based on sequencing efforts of orthologs and the increase in RNA sequence data collection, genes can be mapped to the plant genome of interest. Genedata Selector helps to manage that strain-specific data. Statistical tools and data visualization tools plus integration of different datasets (transcriptomics, metabolomics, and meta data form the public sequence) can all be integrated to describe the strain data (phenotype). Customers want to integrate these disparate datasets despite the different workflows.
In barley, the key goal is to determine how to isolate drought-resistant strains. Genedata Selector enables researchers to manage the sequence data acquisition, process the data to perform sequence annotations in the genome, map phenotypic data, manage strain information, and then profile mutations across different strains to correlate with the phenotypic map.
Genedata Selector also enables researchers to search the identified mutations and use bioinformatics tools to overlay these mutations over the existing patent database to evaluate which ones are free to operate with respect to the gene/mutation (e.g., viable target identification).
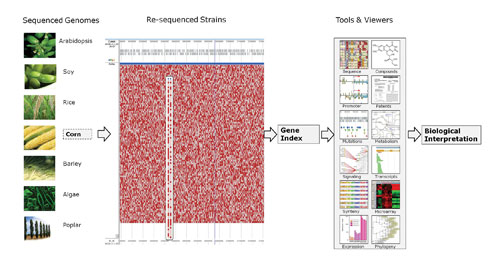
Genedata Selector is an open, integrated, and flexible platform for all omics data. Genome sequences and annotation data are connected to pathways, patent information, and phenotypes.

