
May 1, 2013 (Vol. 33, No. 9)
Comparative genomics is akin to comparing notes across different species. By comparing features at the genetic level—of completely sequenced (or nearly so) genomes—comparative sequence analyses facilitate the functional annotation of genomes and elucidate whole-genome approaches across species.
Recent studies show the difficulty of detecting a disease-causing mutation within a large volume of rare variants in the human population, despite a large number of genomes available for study. Of the 7,098 complete mitochondrial genomes, researchers identified 6,110 single nucleotide variants (SNVs), but could not detect more than 18 known pathogenic mutations.
Comparative genomics is a powerful approach for identifying and interpreting functional elements in the human genome. “Even when the human genome was first sequenced, it was unclear how many protein-coding genes were in it,” says Kerstin Lindblad-Toh, Ph.D., scientific director of vertebrate genome biology at the Broad Institute. “With the sequencing of the second mammalian genome, the mouse genome, it was apparent that 1.3% of genome coded for protein, but that there were many more regulatory elements to find.”
Dr. Lindblad-Toh and colleagues have already reported the sequencing and comparative analysis of 29 eutherian genomes. The high-resolution map of human evolutionary constraint using these mammalian genomes suggests that “at least 6.5% of the human genome has undergone purifying selection.”
“Overlap with disease-associated variants indicates that our findings will be relevant for studies of human biology, health, and disease,” according to Dr. Lindblad-Toh.Researchers have located constrained elements covering almost 4.2% of the genome. Constrained elements are regions of high sequence similarity in the genome across many species.
As Dr. Lindblad-Toh explains, the genomewide studies involving single nucleotide polymorphisms (SNPs) and their association with disease suggest that the 29 mammalian vgenomes will help find disease mutations in the human genome. “More than 85% of signals from genome-wide association studies of human disease fall outside protein-coding genes.”
Dr. Lindblad-Toh and colleagues have employed evolutionary signatures and comparisons with experimental datasets to assign candidate functions for nearly 60% of constrained bases, which “reveal a small number of new coding exons, candidate stop codon read-through events, and over 10,000 regions of overlapping synonymous constraint within protein-coding exons.”
They found “220 candidate RNA structural families, and nearly a million elements overlapping potential promoter, enhancer, and insulator regions.” They have also reported specific amino acids that are positively selected for, 280,000 noncoding elements exapted from mobile elements, and over 1,000 primate- and human-accelerated elements.
Given that 150–200 mammalian genome sequences are necessary to achieve single-base resolution for functional constraint, Dr. Lindblad-Toh’s team has undertaken sequencing of more mammals, including the squirrel monkey, the manatee, the chinchilla, the naked mole rat, the star-nosed mole, and the white rhinoceros. “The main goal is to decipher each and every single base pair in terms of its functional role and identify novel functional elements of significance to human health and disease.”
In a different study, investigators at the Broad Institute of MIT and Harvard used full-genome sequence variation from the 1000 Genomes Project and the comprehensive multiple signals test to probe into 412 candidate selection signals. Further delving into the functional annotation, protein structure modeling, epigenetics, and association studies, they have identified and annotated candidate variants to develop a catalog for experimental follow-up.
Among other variants that include 35 high-scoring nonsynonymous variants and 59 variants associated with expression levels of a nearby coding gene or lincRNA, the researchers identified several mutations linked with susceptibility to infectious disease and other phenotypes.
They have experimentally characterized one candidate nonsynonymous variant in Toll-like receptor 5. Changes in NF-κB signaling are mediated by this variant in response to bacterial flagellin. As Dr. Lindblad-Toh comments, “comparing the hundreds of different mammalian genomes helps in developing models of human disease based on genetic studies and understanding of genome function.”
Rhesus macaques (Macaca mulatta) are proven models for the study of diseases such as HIV/AIDS, cardiovascular disease, obesity and diabetes, asthma, addiction, and age-related diseases of public health significance. “Our research is focused on the development of genetically defined nonhuman primate disease models by characterizing genomic variation between and within nonhuman primate species,” says Betsy Ferguson, Ph.D., associate professor at the neuroscience division of Oregon National Primate Research Center, Oregon Health & Science University.
Dr. Ferguson and colleagues have undertaken comparative analysis of the Indian-origin and Chinese-origin rhesus macaque genomes. The findings reveal variants that suggest currently unrecognized disease susceptibilities in the different populations.
“While they have certain common variants, there may be unique differences that offer an advantage to studying Chinese macaques, e.g., in HIV/AIDS, in that the similarities to human disease progression are much closer,” explains Dr. Ferguson. In contrast, “the Indian rhesus might be more akin to a shorter course of the human AIDS.” On the other hand, the “Chinese species show traits related to a range of anxiety and stress behaviors that mirror those seen in humans.”
For genome variant detection, the investigators used Illumina’s HiSeq platform to achieve deep sequence coverage (30–45x) in an initial six Indian-origin and six Chinese-origin rhesus macaque genomes. BWA aligner methodologies were used to align sequence reads to the rhesus macaque reference genome.
The SAMtools software suite was used to check mapped reads for duplicates and SNVs. “We identified in excess of 19 million SNPs in the rhesus macaque populations,” Dr. Ferguson says. The SNP allele-frequency, potential population-specificity, and the predicted functional effects of the variants are used to identify population differences in risks for disease.
The researchers have successfully evaluated a human exome capture design for the selective enrichment of exonic regions of nonhuman primates including nine chimpanzees, two cynomolgus macaques, and eight Japanese macaques. Their findings indicate that the human exon-capture methods offered an attractive, cost-effective approach for the comparative analysis of nonhuman primate genomes, including gene-based DNA variant discovery. It facilitated efficient enrichment of nonhuman primate gene regions.
The investigators captured over 91% of the target regions in the nonhuman primate samples, although with decreasing specificity as evolutionary divergence from humans increased. They have identified both intraspecific and interspecific DNA variants, validating 85.4% of 41 randomly selected SNPs using Sanger-based sequencing techniques. The findings indicate that a majority (54.6–77.3%) of the variants resulted in a change of three base pairs.
The comparative analysis of functional and nonfunctional variation in rhesus macaque serves as a model for human biology. It sheds light on how variation in population history and size altered patterns and levels of sequence variation in primates. Comparative analyses suggest that rhesus macaque has nearly three times higher SNP density and average nucleotide diversity than humans.
The highest SNP density and average nucleotide diversity were seen in intergenic regions, while the lowest density was observed in the CDS (coding sequences) in both humans and macaques. Indeed, rhesus macaques are almost three times as diverse as the human but more closely equivalent in damaging variation.
Reproductive Biology
Comparative genomic studies with rats and nonhuman primate hypothalamus suggest implications for reproductive physiology. Using targeted delivery of RNA interference, to either disrupt (rats) or abolish (monkeys) reproductive cycles by selective inhibition of enhanced at puberty 1 (EAP1) expression in discrete regions of the hypothalamus, researchers found that menstrual cycles went haywire when hypothalamic EAP1 gene expression was blocked. This suggests that diminished EAP1 function may contribute to disorders of the menstrual cycle of neuroendocrine origin. Polymorphisms in EAP1 may increase the risk of functional hypothalamic amenorrhea in humans.
It is well known that female reproductive capacity and the maintenance of mature reproductive function are under transcriptional control of specific gene networks operating within the neuroendocrine brain. Dr. Ferguson and her team showed that a single-nucleotide polymorphism in the 5´-flanking region of EAP1 gene resulted in an increased incidence of amenorrhea/oligomenorrhea in nonhuman primates. “It is a beautiful study with significant findings illustrating epigenetic modification, a first of its kind from nonhuman primates with implications for human disease,” says Dr. Ferguson.
Identifying the natural genetic variations that underlie complex disease traits will enhance the translational and pharmacogenomic value of nonhuman primate models such as rhesus macaques, says Dr. Ferguson.
Compared with the comprehensive evolutionary approaches of the investigators at the Broad Institute, involving hundreds of different mammalian genomes, Dr. Ferguson is focused on digging into the sequence variation in nonhuman primate species to maximize their translational value in developing tractable models that parallel common human disease.
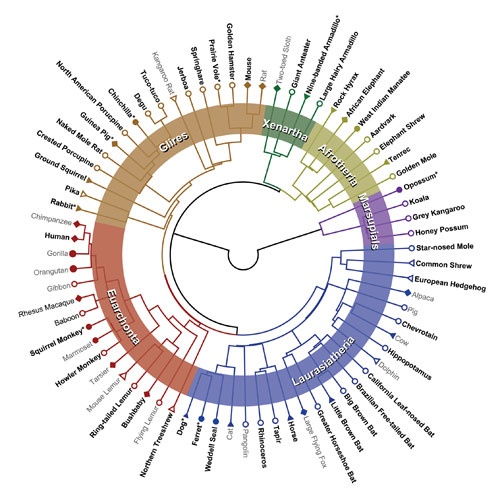
Representatives of the more than 70 mammals sequenced or in the sequencing queue to help understand the human genome: Comparison of mammalian genomes helps decipher the function of genes and regulatory elements, and can guide studies of human disease. [Broad Institute]
Sequencing Technologies Hold the Key
Different DNA sequencing technologies are used to sequence a human genome. Sequencing of clinical samples is driven by research requirements such as urgency, depth of coverage, and budget. Competing technologies are available—from Illumina, Complete Genomics, Life Technologies’ SOLiD, or emerging technologies such as Ion Proton or tools from Pacific BioSciences.
The key facets of genome sequencing analysis include: SNVs, copy-number variations (CNVs), more complex structural variants, and the regions showing loss-of-heterozygosity (LOH). Clinical and biological researchers apply these data types regularly to arrive at decisions, such as determining cancer treatment or characterizing patterns of genetic variation. In addition, it can be used to highlight clinically actionable genetic variants for purposes of molecular ancestry and comparative genomics, or highlight known drug metabolism variants.
“Regardless of the sequencing platform selected for the genome analysis, there is an urgent need to convert the sizeable volumes of digital data into an accessible format that can be used to direct subsequent research,” says Stephen Rudd, Ph.D., formerly with the Malaysian Genomics Resource Centre Berhad (MGRC), and currently head of computational biology at QFAB, Institute for Molecular Biosciences, Queensland Bioscience Precinct, The University of Queensland.
As yet, there is no ideal single workflow that performs all of the analyses in a streamlined fashion that is accessible to a researcher with limited bioinformatics expertise. The MGRC set out to address this dearth of researcher-friendly solutions by implementing a portable application pipeline that could be deployed close to the researcher.
“The One-Click Pipeline and Hotspot data presentation layer are part of MGRC’s portfolio of genome bioinformatics tools and are used to present content to clinical researchers, biologists, and support bioinformaticians,” says Dr. Rudd.
Genome One-Click entails application of a sequential pipeline of sequence transformations. Stringent quality control ensures that reads are mapped against the reference human genome using software such as SXMapper. The mapping process helps to understand the key metrics of variation detection, such as uniqueness of the sequence context, the local repetition, and depth of coverage.
Subsequent iterative steps help to characterize possible variants and to assign suitable parameters for the variation assignment. The results from the Genome One-Click are available as text files or BAM files. The technology can typically map a human genome sequenced to 50x coverage, with variations determined within 48 hours.
“At the heart of the One-Click Genome analysis pipeline is MGRC’s genome mapping software, built upon the core Synasuite set of bioinformatics tools and delivering a high-performance genome analysis without the need for computer clusters and oversized data centers,” explains Dr. Rudd.
Hotspot is a mutation-mining framework used to explore and characterize genome-scale data by curating the Genome One-Click results into a relational data structure. According to Dr. Rudd, “our one-click pipeline has mapped tens of pairs of genomes to the reference human genome and the resulting mapping data is stored in the Hotspot system.” Using password-protected online access to the genome data and a set of filters and controls, candidate features can be queried.
For example, queries can be built to identify novel SNPs located in miRNA genes in tumor samples but not in their paired normal samples. Or in other cases, experts can determine nonsynonymous protein substitutions that segregate between Chinese and Malay individuals, for instance. The variants within the databases acquire the confidence scores calculated by Genome One-Click. The results can be filtered for the strict canonical and ideal variants, or relaxed to accommodate some of the more speculative content, Dr. Rudd explains.
Analysis of a human genome and the required mapping of hundreds of millions of short sequence reads require a significant amount of computing power. The transfer of raw sequencing data can also be a challenge for an effective and fast turnaround time. Comparison of the software with other mapping software and variant-calling methods has shown that the MGRC approach to the analysis of whole-genome data is robust, cost-effective, rapid, and attended with good sensitivity and selectivity, says Dr. Rudd.
“It is especially useful for researchers who need to extract the essence from multiple human genomes or exomes but are not familiar with bioinformatics and the plethora of complex and not always interoperable open source solutions,” he says.
At MGRC, experts plan an integrated data analysis environment that could be provided globally—either as a cloud solution in a centralized data center, or as a standalone appliance that could be deployed in a hospital, university, or pharmaceutical company data center, according to Dr. Rudd. “Given that human genome sequencing can generate terabytes of data, our customers find it more convenient to ship us a hard disk drive, which we then return to them with the results of the analysis.”
The One-Click Pipeline and Hotspot were applied to the Malaysian MyGenome project involving 26 deep sequenced human genomes, delivering the first systematic survey of human genetic variation to the Malaysian Ministry of Science Technology and Innovation. Currently, the software suite is being used in projects offering genomics services for clinical and pharmaceutical customers in Europe and the U.S. to interpret the extensive data emerging from whole-genome sequencing.

