
In prokaryotes, transcription is a significant step of regulation for gene expression, and the precise identification of transcriptional landmarks such as transcription start sites (TSS) and termination sites (TTS) is critical for understanding microbial responses to perturbation. Compared to eukaryotes, prokaryotic transcripts do not have introns and consequently, have a linear correspondence to their genomic locus. Despite this apparent simplicity, prokaryotic transcriptome studies are unexpectedly challenging: Bacteria can vary the gene content of their transcripts by using alternative TSS and TTS, resulting in several overlapping transcripts reminiscent of alternative splicing. For example, on a single E. coli locus, up to 15 overlapping transcripts with distinct gene content have been reported1.
Furthermore, processed RNA (such as rRNA and tRNA) accounts for more than 95% of total RNA in the prokaryotic cell2, and mRNA enrichment is essential for meaningful coverage of mRNA. Moreover, the turnover of bacterial mRNAs can be remarkably rapid (the average half-lives of mRNA is in the range of 2-10 min3), therefore transcripts that are being degraded accounts for a large fraction of total RNA masking notably the internal TSS and TTS.
Thus, for the accurate representation of primary transcriptomes, it is crucial to distinguish only transcripts that have retained their original 5ʹ and 3ʹ ends from the myriad of other RNA molecules: this strategy has the double advantage of removing the rRNA and defining TSS and TTS. Yet, this task has also proven difficult: indeed, prokaryotic mRNAs lack the signature 3ʹ end polyA tail and the 5ʹ Cap structure typically used as handles to ensure sequencing of the desired RNA in eukaryotes.
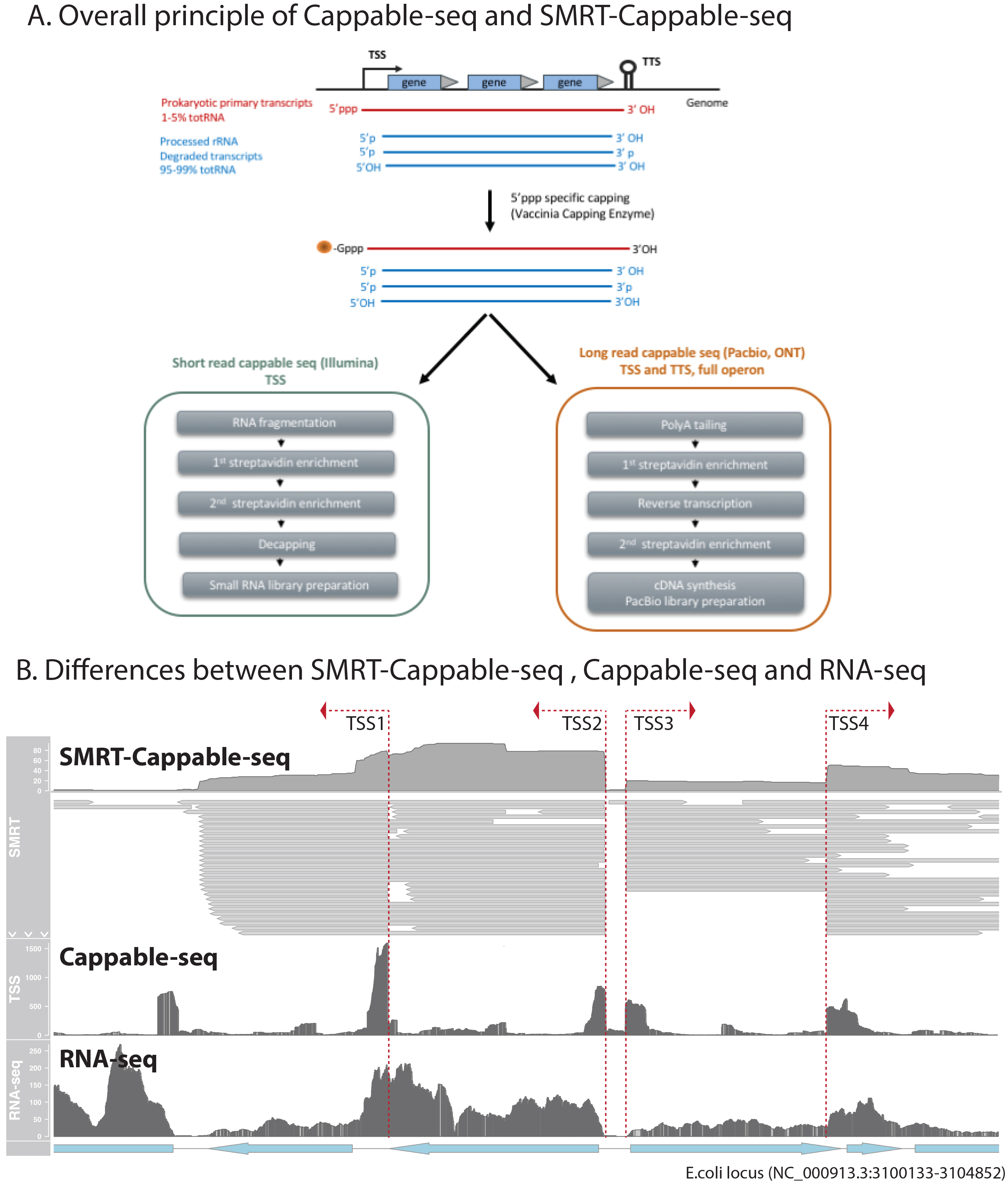
Here we report an advanced method called Cappable-seq5 to specifically label the 5ʹ end and therefore isolate the prokaryotic primary transcripts. Combined with Illumina short read sequencing, Cappable-seq offers the ability to investigate prokaryotic TSS at single-base resolution5 (Figure 1A). SMRT-Cappable-seq 1 (Figure 1A) uses long read sequencing to provide contiguous sequencing of full-length primary transcripts suitable for genome-wide identification of prokaryotic operon structure. Figure 1B shows the TrmI-1 locus in E. coli illustrating the distinctive results obtained using Cappable-seq, SMRT-Cappable-seq and the widely used RNA-seq.
General Principle of Cappable-seq
The first step of most in vivo RNA degradation pathways in bacteria is believed to be the removal of the triphosphate present on the 5ʹ nucleotide of primary transcripts. Processing of RNA such as rRNA maturation leaves a 5ʹOH or a 5ʹ monophosphate. Thus, primary transcripts can be differentiated from other RNAs based on their 5ʹ end. This molecular distinction between primary and processed transcripts forms the basis of Cappable-seq5. Cappable-seq relies on the vaccinia capping enzyme (VCE, NEB, M2080) to specifically cap the di- or triphosphorylated 5ʹ end of a primary transcript with a biotin-derived cap. These capped RNAs can be captured explicitly via a streptavidin bead system, allowing to isolate the primary transcripts while removing uncapped RNAs individually.
Method and consideration
Both Cappable-seq and SMRT-Cappable-seq methods are based on the same principle (workflows shown in Figure1A). However, these methods differ according to the sequencing platforms and objectives:
• Cappable-seq is developed for short-read Illumina sequencing, and the RNA is fragmented prior to streptavidin enrichment leading to the selection of the most 5ʹ end fragment of the primary RNA. Combined with the small RNA library preparation (NEB, E7330), the resulting library can be sequenced using Illumina to identify TSS at nucleotide and strand resolution. Short read offers the possibility of sequencing at high-throughput resulting in a good quantification of the TSS usage (Figure 2A). Furthermore, each transcript can be summarized to a single sequencing tag enabling digital gene expression analysis of the 5ʹ end. TSS identified by Cappable-seq allows to precisely locate and study the promoter, but gene assignment can be difficult. Indeed, TSS are generally found in intergenic regions, and transcripts in bacteria are often polycistronic. Starting material for Cappable-seq ranges from 2-5 µg of total RNA.
•For SMRT-Cappable-seq, the enrichment is done on intact RNA and utilizes long-read sequencing platforms such as Pacbio or Oxford Nanopore (ONT-Cappable-seq) to obtain full-length transcripts. Since the SMRT-Cappable-seq is designed to capture full-length transcripts containing both TSS and TTS, it requires the isolation of intact high-quality primary transcripts. RNA extraction is, therefore, a critical step in the procedure. The RNA is subsequently capped, polyA-tailed, and enriched with streptavidin before it goes through reverse transcription, a second enrichment step, PCR amplification and finally, Pacbio library preparation. After PCR amplification, replacing the PacBio SMRT-bells with Nanopore adaptor permits ONT-nanopore sequencing instead.
Results
A. Cappable-seq
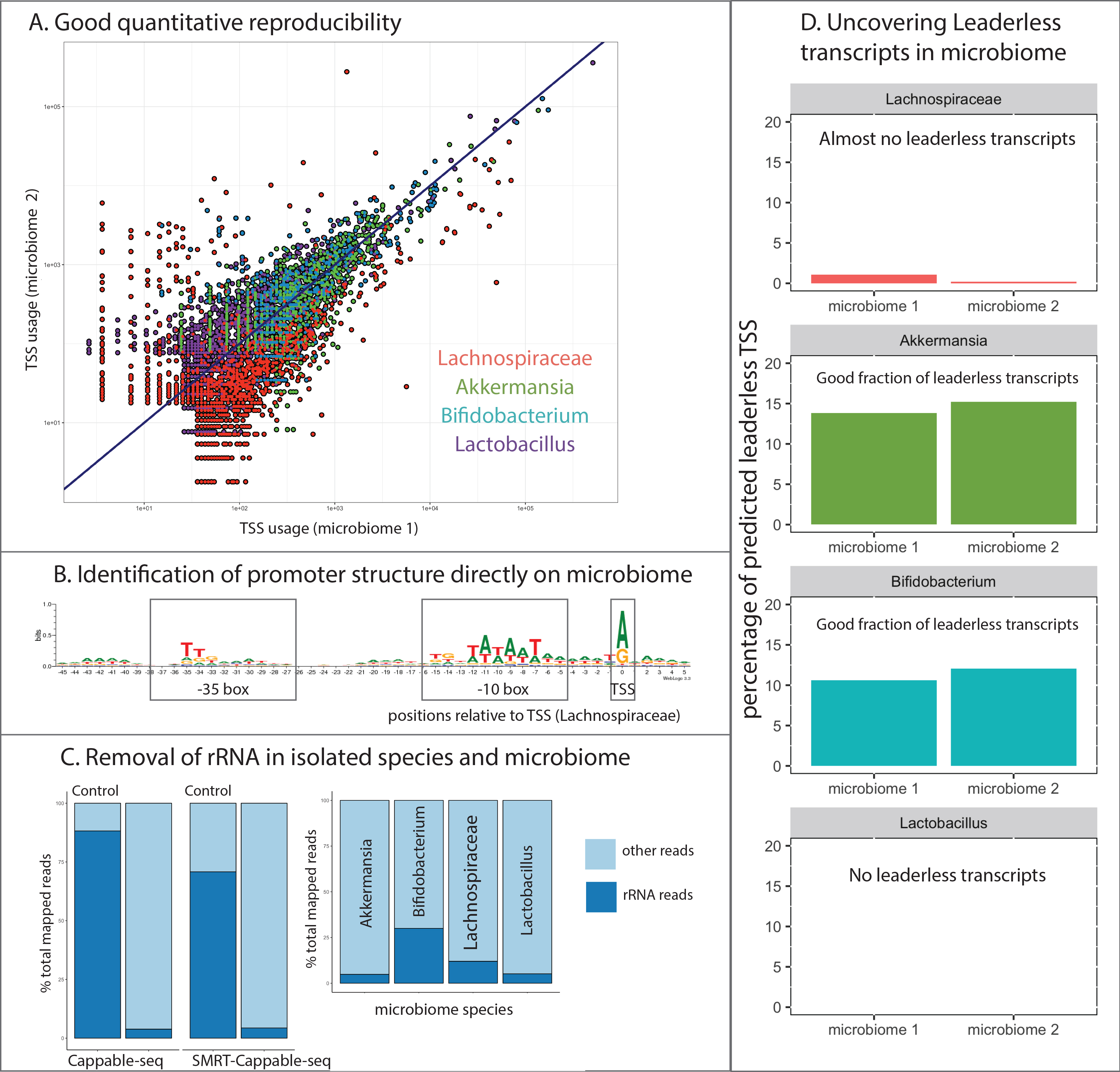
Applied to E. coli, Cappable-seq identifies TSS at nucleotide and strand resolution and removes processed ribosomal RNA. On a standard Cappable-seq library, we routinely get only 4 % ribosomal RNA remaining (Figure 2C). Cappable-seq libraries can be complemented with a control library for which the streptavidin enrichment step has been omitted. Similar to differential RNA-seq (dRNA-seq), comparing the Cappable-seq with the control library, offers the possibility of identifying highly confident TSS. From approximately 20 million reads, Cappable-seq identifies around 16,000 highly confident TSS clusters detecting 76% of all E. coli genes. Thus, the throughput of a Miseq run is enough for defining the landscape of promoter in a single bacterium.
Similarly, Cappable-seq can be applied to a microbiome. As illustrated using four representative species present in the microbiome of a mouse gut, only a minority of reads are mapped to ribosomal genes (Figure 2C). TSS were found at single-base resolution highlighting the promoter configuration of the species studied (Figure 2B). Interestingly Cappable-seq also uncovers putative alternative mode of transcription such as leaderless transcription (Figure 2D). Importantly, a genomic reference sequence is required to associate TSS with their cognate genes and promoters.
B. SMRT-Cappable-seq
SMRT-Cappable-seq has a reasonably good correlation with Illumina RNA-seq, allowing for quantification of transcripts while removing most of the ribosomal RNA (Figure 2C). Nonetheless, the strength of this method lies on the ability to identify the rich landscape of full-length transcripts that are often found overlapping due to alternative usage of TSS and the read-through of TTS1. Applied to E. coli, this technology results in an accurate definition of the transcriptome with 34% of known operons from RegulonDB being extended by at least one gene. Furthermore, 40% of transcription termination sites have read-through that alters the gene content of the operons1. Applied to a microbiome, SMRT-Cappable-seq is expected to identify full-length transcripts revealing the operon structures across a diverse population of bacteria. Identification and annotation of open reading frames can be theoretically done directly on reads without the requirement of assembled reference genomes.
References
- Yan B, Boitano M, Clark TA, Ettwiller L. SMRT-Cappable-seq reveals complex
operon variants in bacteria. Nat Commun. 2018, 9:3676. - Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation,
physiology and pathogenicity. Nat Rev Genet. 2010 11:9-16. - Laalami S, Zig L, Putzer H. Initiation of mRNA decay in bacteria. Cell Mol Life Sci 2014, 71:1799-1828.
- Jäger D, Förstner KU, Sharma CM, Santangelo TJ, Reeve JN. Primary
transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genomics 2014, 15:684. - Ettwiller, L., Buswell, J., Yigit, E. & Schildkraut, I. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single-base resolution in a model prokaryote and the gut microbiome. BMC Genomics 2016, 17:199.
Bo Yan, PhD, is a research scientist at New England Biolabs, Chloé Baum is a doctoral candidate at New England Biolabs and Genoscope (France), and Laurence Ettwiller, PhD, is a Senior Scientist in the research department at New England Biolabs.

