
May 15, 2013 (Vol. 33, No. 10)
Molecular techniques and technologies to probe alterations in gene and protein expression that may contribute to the development, progression, prognosis, heterogeneity, therapeutic options, and drug-resistance of cancers and tumor types are an increasingly important part of the clinician’s diagnostic toolbox.
Researchers and clinicians alike are relying on sequencing technology and genomic, proteomic, and metabolomic strategies to identify biomarkers with the potential for use in diagnostics, drug discovery and development, and patient stratification for clinical drug testing.
Commenting on the recent AACR meeting, analysts from Mizuho Securities observed that molecular diagnostics are “clearly an essential part of oncology,” with “a lot of optimism around cancer genome sequencing, even if most attendees do not think it will replace existing diagnostics in the near future.” They report “significant need for better biomarkers and companion diagnostics,” with biomarkers and diagnostics “being used earlier in development, but still under-utilized.” Additionally, “genomics is clearly driving many aspects of oncology R&D,” the Mizuho analysts explained.
Several presentations at the AACR meeting highlighted the application of molecular diagnostic tools in cancer R&D and product development for the oncology diagnostics market.
Mirza Peljto and co-authors from Flagship Biosciences and Affymetrix/Panomics described a method for the quantitative in situ assessment of oncogene RNA and protein expression in human clinical tumor tissue for use in biomarker and diagnostics development. In breast cancer, the Her2 oncogene is amplified or overexpressed in about 30% of breast cancers, but the correlation between Her2 gene expression and protein expression in these tumors is not well understood.
The researchers used chromogenic RNA in situ hybridization (CISH) and immunohistochemistry (IHC) to develop a quantitative image analysis-based assay in which the levels of Her2 protein and RNA can be compared in situ within tissue context. This approach allows for the measurement and correlation of Her2 RNA and membrane protein expression across an entire tissue section. “It improves diagnostic concordance by relying on an automated method and also improves confidence in interpretation by assessing every tumor cell across the whole slide,” said Peljto.
The authors were able to distinguish high- from low-expressing biopsy samples using the CellMap™ algorithm to analyze the digital images produced, to differentiate tumor cells from the normal surrounding tissue, and to transform the images into cell and biomarker maps and quantify the levels of RNA and protein biomarkers.
The authors reported 79% concordance between Her2 RNA and Her2 protein expression. The study results “suggest the potential use of RNA CISH in assessment of Her2 status in conjunction and/or parallel to IHC.” The authors drew the following conclusions: “We demonstrated practical feasibility of combined molecular and image analysis for analyzing clinical tumor samples. The inclusion of automated, whole-slide IHC and CISH interpretation for molecular assessment for companion diagnostics may help solve the long-standing problem of manual pathologist assessments and improve concordance with the inclusion of an RNA-based readout simultaneous to an IHC-based readout.”
A visual representation of how CellMap distinguishes and quantifies Her2 membrane staining and Her2 RNA CISH in whole slides of clinical tissue TMAs: CellMap defines and quantifies membranes even with relatively high cytoplasmic background staining. (A) original IHC image; (B) CellMap algorithm- generated markup for IHC analysis (red colored membranes indicate cells with high expression of Her2 while orange colored membranes indicate medium levels of Her2 expression). Using algorithm-based imaging it is possible to quantify RNA CISH for Her2. CellMap identifies individual cells as well as dots containing Her2 RNA from CISH signals based on the size of individual dots as well as their signal intensity. (C) Original CISH image; (D) CellMap algorithm-generated markup image of CISH analysis. The cells are binned into high expressing (red; >7 dots), medium expressing (orange; 5–7 dots), low expressing (yellow; 3–5 dots), negative (blue;
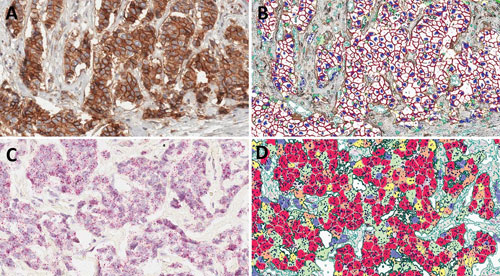
CellMap in action. [Flagship Biosciences]
DNA and Protein Biomarker Analysis
Predictive Biosciences has developed commercial cancer diagnostic tests based on its Clinical Intervention Determining Diagnostic (CIDD)™ approach. The CIDD method uses a multi-analyte diagnostic readout based on measures of both DNA and protein markers. The dual cutoffs allow for >90% assay sensitivity and specificity.
At AACR, company representatives spoke about the detection of bladder cancer-associated mutations and epigenetic changes.
Instead of yielding a positive/negative result, the approach enables the stratification of symptomatic populations into three groups based on the likelihood of cancer: high likelihood of cancer, which identifies candidates for maximum, accelerated therapeutic intervention; possibility of cancer, a middle group of patients who should receive the standard of care; and high likelihood to be disease-free, or patients that do not require further evaluation or treatment.
“The addition of DNA biomarkers that have high specificity, to protein biomarker-based diagnostic testing, essentially shifts the DNA marker-positive samples to the high-risk group, targeting those patients for intervention,” said Cecilia Fernandez, director of applied science. “The increased sensitivity of the multi-analyte assay allows for a higher protein marker cutoff to be set and a larger proportion of patients to be characterized as disease-free based on urine testing, sparing them further evaluation and invasive tissue sampling.”
This concept was also described by John Millholland from Predictive Biosciences. His presentation focused on research aimed at applying a next-gen, deep amplicon sequencing method that can achieve quantitative gene mutation detection when mutations are present at levels as low as 0.02% of the normal DNA, as might be the case if testing were performed using urine instead of a bladder tissue sample.
The authors show that compared to qPCR, deep amplicon sequencing of the FGFR3 gene yielded significantly greater concordance in mutation detection in tissue vs. urine—90% concordance with sequencing vs. about 50% with qPCR. “The additional mutations detected in urine using deep sequencing vs. in tissue samples (15 of 19 urine samples vs. 11 of 19 urine samples with qPCR) suggest that urine might be more representative of the entire organ versus tissue sections that might have stochastic sampling of the tumor,” Millholland and his co-authors reported.
When the researchers added a second set of bladder cancer-associated mutations to the assay, these in the TP53 gene—associated with invasive bladder cancer, as opposed to the link between FGFR3 mutations and noninvasive disease—assay performance improved. They reported no overlap between samples positive for FGFR3 and TP53 mutations. The mutations in the two genes complement each other in the assay, and the combinations increases the sensitivity of the assay. “By combining TP53 with our current FGFR3 assay, we can provide excellent detection of all stages and grades of bladder cancer,” said Anthony P. Shuber, CTO.
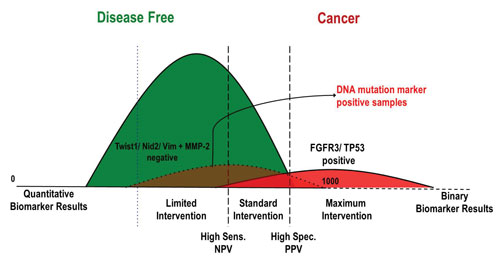
Multi-analyte diagnostic readout (MADR): The use of quantitative markers, such as protein markers, can result in very high sensitivity, but often low specificity (blue dotted line). The addition of binary DNA mutation markers that have very high specificity provide a boost in sensitivity, such that quantitative marker cutoffs can be adjusted to increase specificity (black dashed lines). [Predictive Biosciences]
In a third presentation from Predictive Biosciences, researchers also described a technique developed to detect hypermethylated genes in bodily fluids such as urine. They have incorporated into the multi-analyte diagnostic readout assay described above a next-gen bisulfate sequencing approach to determine the methylation of CpG sites at single-base resolution.
Identification of altered methylation patterns is an increasingly valuable diagnostic tool in cancer. The technique described here “could be used to detect other hypermethylated genes in a variety of bodily fluids where percent methylation may be much lower than in tumor tissue samples,” concluded the authors. “By developing a version of the assay that integrates all of the biomarkers in a format that can be run on any NGS platform, we can increase throughput, reduce cost, and make the assay ready for in vitro diagnostic kit development,” explained Shuber.
Nicola Normanno, Centro Richerche Oncologiche Mercogliano (Italy), and an international team of colleagues that participate in the OncoNetwork Consortium, evaluated a custom gene panel that targets hotspot mutations in 22 genes implicated in colorectal cancer and non-small-cell lung carcinoma. The panel is intended for use in a clinical research setting to help classify colon and lung tumors based on the detection of cancer-associated gene mutations.
Normanno et al., described the three-phase study the consortium used to verify the panel using 155 FFPE tissue samples and controls on an Ion Torrent 316 chip.
Ten ng of sample were processed on the OncoNetwork panel following amplification with the Ion AmpliSeq Library Kit 2.0. The authors reported high average per base accuracy of the panel, detection of all expected variants, a 100% true positive rate for newly identified variants as confirmed using Sanger sequencing, and 100% reproducibility of results across six different testing laboratories. A second gene panel, designed to optimize read distribution over the amplicons, was introduced during the course of the three-phase verification study, and phases 1 and 2 of the study were repeated using the new panel design. It allowed for multiplexing of up to eight samples on a 316 chip with an average read depth of 2,750 and improved coverage uniformity. Repeat testing confirmed 100% reproducibility and high genotyping sensitivity of the new panel.
Colorectal Cancer
Todd Hembrough, Ph.D., and colleagues from OncoPlex Diagnostics and the University of Chicago described their work to develop a multiplexed mass spectrometry-based quantitative assay for colorectal cancer (CRC) using liquid tissue-selected reaction monitoring (SRM). The assay is based on an understanding of the relationship between overexpression of multiple oncogenes, including those encoding several families of receptor tyrosine kinases, and tumor heterogeneity.
This link allows for characterization of protein expression by targeted proteomics in CRC tumors to categorize patients into molecular subsets and identify a personalized treatment strategy. “Moreover, intra-patient tumor heterogeneity from primary tumor to metastatic disease is likely to influence biomarker prediction of response to specific targeted agents,” stated the researchers.
They validated the CRC-plex SRM assay preclinically on cell lines, and demonstrated a good correlation between Liquid Tissue®-SRM analysis and ELISA for quantification of selected oncogene proteins. In this study the authors measured oncogene protein levels using SRM analysis in 42 CRC-paired primary and metastatic tumor tissue samples from the same patients. They were able to measure as many as 20 target proteins in a multiplexed assay using FFPE tumor tissue samples.
A comparison of primary CRC tumors with their paired metastatic samples showed a statistically significant increase in cMet expression for metastatic vs. primary tissues, “suggesting that in the absence of gene amplification, cMet may be a druggable target in these tissues,” concluded the authors.
Leveraging Molecular Techniques
A team of researchers from MolecularMD was focused on PTEN, a tumor suppressor that exerts its effects via dephosphorylation. Loss of PTEN function has been associated with multiple cancers, including endometrial cancer, glioblastoma, melanoma, and prostate cancer. Loss of PTEN activity can result from more than one independent aberration, including mutations in the coding regions (exons) of the PTEN gene, genomic deletions, or promoter methylation, in which the PTEN gene may be present and unadulterated but epigenetic changes to the regulatory DNA upstream of the gene block expression, resulting in loss of the PTEN protein.
The researchers from MolecularMD evaluated FFPE tissue samples using three different methods, allowing them to assess PTEN status at both the DNA and protein levels. Both “may be of important for clinical decision-making because loss of PTEN is associated with resistance to anti-EGFR therapies,” they explained.
Using a Sanger sequencing assay on genomic DNA extracted from FFPE tissue sections and amplified with PCR they could detect known hot-spot mutations in specific exons. The automated immunohistochemistry assay they developed was validated in cell lines and FFPE tissues. If >10% of the tumor cells in a sample stained, the sample was defined as positive for PTEN protein. The two-color dual chromogenic and silver in situ hybridization (ISH) assay was scored as follows: >20% loss of PTEN signal identifies tumors having heterozygous loss of the PTEN gene; >30% loss of PTEN signal indicates homozygous loss of the PTEN gene.
Assessment of 22 FFPE tissue specimens showed that of the nine samples identified as wild-type by Sanger sequencing, seven had normal PTEN protein expression on IHC. The fact that two samples were IHC-negative suggests that mechanisms other than mutations in the coding regions of the PTEN gene are responsible for the loss of PTEN protein. Of the 13 samples found to have hot-spot mutations on Sanger sequencing, seven showed loss of PTEN protein on IHC; the other six samples had normal PTEN protein expression.
This finding also supports the observation that mutations in gene coding region hot-spots are not sufficient on their own to predict PTEN protein expression, and vice versa. The authors concluded that complementary molecular diagnostic methods that assess the mechanisms underlying changes in gene and protein expression are needed for accurate clinical assessment of PTEN status in FFPE tissue samples.

