
Anna Louise Swan
Ali Mobasheri
David Allaway
Susan Liddell
Jaume Bacardit
A branch of artificial intelligence could be used for identifying biomarkers and classifying samples into disease or treatment groups.
Abstract
Mass spectrometry is an analytical technique for the characterization of biological samples and is increasingly used in omics studies because of its targeted, nontargeted, and high throughput abilities. However, due to the large datasets generated, it requires informatics approaches such as machine learning techniques to analyze and interpret relevant data. Machine learning can be applied to MS-derived proteomics data in two ways. First, directly to mass spectral peaks and second, to proteins identified by sequence database searching, although relative protein quantification is required for the latter. Machine learning has been applied to mass spectrometry data from different biological disciplines, particularly for various cancers. The aims of such investigations have been to identify biomarkers and to aid in diagnosis, prognosis, and treatment of specific diseases. This review describes how machine learning has been applied to proteomics tandem mass spectrometry data. This includes how it can be used to identify proteins suitable for use as biomarkers of disease and for classification of samples into disease or treatment groups, which may be applicable for diagnostics. It also includes the challenges faced by such investigations, such as prediction of proteins present, protein quantification, planning for the use of machine learning, and small sample sizes.
Introduction
The comparison of samples belonging to different physiological states is vital in the search for putative biomarkers, and proteomics provides suitable methods for this purpose, through the quantitation of proteins. Proteomics provides some advantages over transcriptomics as it can both be used in cell-free biological fluids, such as serum, urine, and synovial fluid, and provide further knowledge such as through post-translational modifications. Quantitative methods, to identify the amounts of proteins, can also be applied, which can be an advantage over quantifying levels of gene expression, depending on the purpose of the study, as gene expression does not necessarily correlate with protein levels. However, the value of this technology is dependent on the quality of the analysis methods used to process the generated data (Bantscheff et al., 2007, 2012).
Machine learning techniques have been utilized broadly to analyze data from many areas of biology; in particular, various machine learning methods have been applied to data generated by the analytical techniques of transcriptomics and metabolomics for classification of unknown samples and identification of genes relevant to the disease state. Similar methods are now being applied to the field of proteomics and, more specifically, the analysis of data generated from tandem mass spectrometry (Sun and Markey, 2011).
There are numerous technologies used to extract quantitative protein information from biological samples. These techniques cover a broad spectrum of approaches, balancing throughput (one/many proteins at a time) and quality of the extracted data.
Commonly used techniques include two-dimensional gel electrophoresis, enzyme-linked immunosorbent assays (ELISAs), protein arrays, affinity separation, and mass spectrometry-based technologies (Ray et al., 2011; Schiess et al., 2009).
A number of these methods, including gels and ELISAs, are limited in the number of proteins they can analyze because of time requirements. They also require specific proteins of interest to be chosen when designing the study and suitable cross-reactive antibodies to be available; this can be challenging for non-model organisms. In comparison, mass spectrometry (MS) techniques can be used as a high-through-put discovery based method; lists of proteins can be identified from samples that are analyzed (Perkins et al., 1999). This means that tandem mass spectrometry can be used to find proteins that may not have previously been considered, provided the proteins can be found within protein sequence databases. A combination of multiple proteomics methods can also be utilized to form an effective analysis pipeline. Ray et al. (2011) showed that MS, in various guises, has been pivotal to biomarker discovery for a range of different diseases. This review will discuss the applications of machine learning for analysis of proteomic mass spectrometry data and the challenges involved. MS-specific challenges, including the identification of proteins using sequence database searching software and protein quantitation or pre-processing for peak analysis, will be covered. As will machine learning-specific considerations, such as the small numbers of samples that can often result from an MS investigation, and the types of machine learning most suited for the required task, either sample classification or biomarker identification. A survey of articles involving the combination of mass spectrometry and machine learning will be followed by a brief discussion of post-machine learning analysis, including literature mining and pathway analysis. An overview of the areas to be covered in this review is provided in Figure 1.
To read the rest of this article, CLICK HERE.
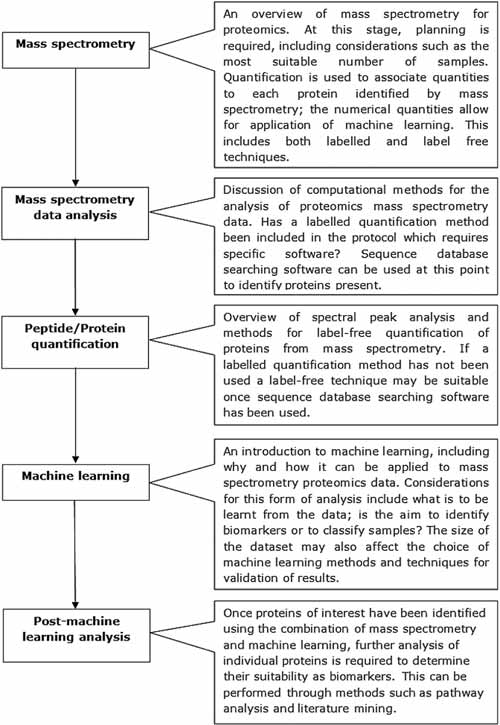
FIG. 1. An overview of the topics covered in this review, including the general work flow required and the major considerations that are necessary before beginning an investigation combining mass spectrometry and machine learning.
Anna Louise Swan is affiliated with the School of Biosciences, Faculty of Science, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom.
Ali Mobasheri is affiliated with the Musculoskeletal Research Group, School of Veterinary Medicine and Science, Faculty of Medicine and Health Science, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom; the D-BOARD European Consortium for Biomarker Discovery, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom; the Arthritis Research UK Centre for Sport, Exercise and Osteoarthritis, Nottingham University Hospitals, Nottingham, United Kingdom; the Arthritis Research UK Pain Centre, The University of Nottingham, Queen’s Medical Centre, Nottingham, United Kingdom; the Medical Research Council and Arthritis Research UK Centre for Musculoskeletal Ageing Research, The University of Nottingham, Queen’s Medical Centre, Nottingham, United Kingdom; the Center of Excellence in Genomic Medicine Research (CEGMR), King Fahad Medical Research Center (KFMRC), King AbdulAziz University, Jeddah, Kingdom of Saudi Arabia; and the School of Life Sciences, University of Bradford, Bradford, United Kingdom.
David Allaway is affiliated with the WALTHAM® Centre for Pet Nutrition, Waltham-on-the-Wolds, Melton Mowbray, Leicestershire, United Kingdom.
Susan Liddell is affiliated with the School of Biosciences, Faculty of Science, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom; the Proteomics Laboratory, School of Biosciences, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom; and the D-BOARD European Consortium for Biomarker Discovery, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom.
Jaume Bacardit is affiliated with the School of Computer Science, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom; and the D-BOARD European Consortium for Biomarker Discovery, University of Nottingham, Sutton Bonington Campus, Leicestershire, United Kingdom.
References:
Bantscheff M, Schirle M, Sweetman G, Rick J, and Kuster B. (2007). Quantitative mass spectrometry in proteomics: A critical review. Anal Bioanal Chem 389, 1017–1031.
Bantscheff M, Lemeer S, Savitski M, and Kuster B. (2012). Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal Bioanal Chem 404, 939–965.
Sun CS, and Markey MK. (2011). Recent advances in computational analysis of mass spectrometry for proteomic profiling. J Mass Spectrom 46, 443–456.
Ray S, Reddy PJ, Jain R, Gollapalli K, Moiyadi A, and Srivastava S. (2011). Proteomic technologies for the identification of disease biomarkers in serum: Advances and challenges ahead. Proteomics 11, 2139–2161.
Schiess R, Wollscheid B, and Aebersold R. (2009). Targeted proteomic strategy for clinical biomarker discovery. Mol Oncol 3, 33–44.
Perkins DN, Pappin DJC, Creasy DM, and Cottrell JS. (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567.
OMICS: A Journal of Integrative Biology, published by Mary Ann Liebert, Inc., is the only peer-reviewed forum covering all trans-disciplinary OMICS-related areas such as integrative (systems) biology and medicine. The above article will be published in the December 2013 issue of OMICS with the title “Application of Machine Learning to Proteomics Data: Classification and Biomarker Identification in Postgenomics Biology”. The views expressed here are those of the authors and are not necessarily those of OMICS: A Journal of Integrative Biology, Mary Ann Liebert, Inc., publishers, or their affiliates. No endorsement of any entity or technology is implied.

