

For decades, Adrian Krainer, professor at Cold Spring Harbor Laboratory (CSHL), has studied the mechanisms of RNA splicing, how they go awry in cancer and genetic diseases, and how to correct faulty splicing. Krainer’s lab has found that it is possible to stimulate protein production by altering mRNA splicing through the introduction into cells of chemically modified pieces of RNA called antisense oligonucleotides (ASOs).
In late 2016, one such molecule, nusinersen (sold by Biogen under the brand name Spinraza), became the first FDA-approved drug to treat spinal muscular atrophy (SMA) by injection into the fluid surrounding the spinal cord. Nusinersen was conceived and tested over several years in SMA mouse models by Krainer and his CSHL colleagues, in a long-standing collaboration with drug developers led by Frank Bennett PhD at Ionis Pharmaceuticals.
Two years earlier, in 2014, Krainer co-founded Stoke Therapeutics (Nasdaq: STOK) with Isabel Aznarez, PhD, to use their groundbreaking science targeting pre-mRNA splicing to develop precision medicines that treat genetic diseases. Headquartered in Boston, Stoke Therapeutics is a biotechnology company focused on upregulating protein expression with RNA-based medicines. Using a proprietary TANGO (Targeted Augmentation of Nuclear Gene Output) approach, Stoke is developing ASOs to restore protein levels selectively.
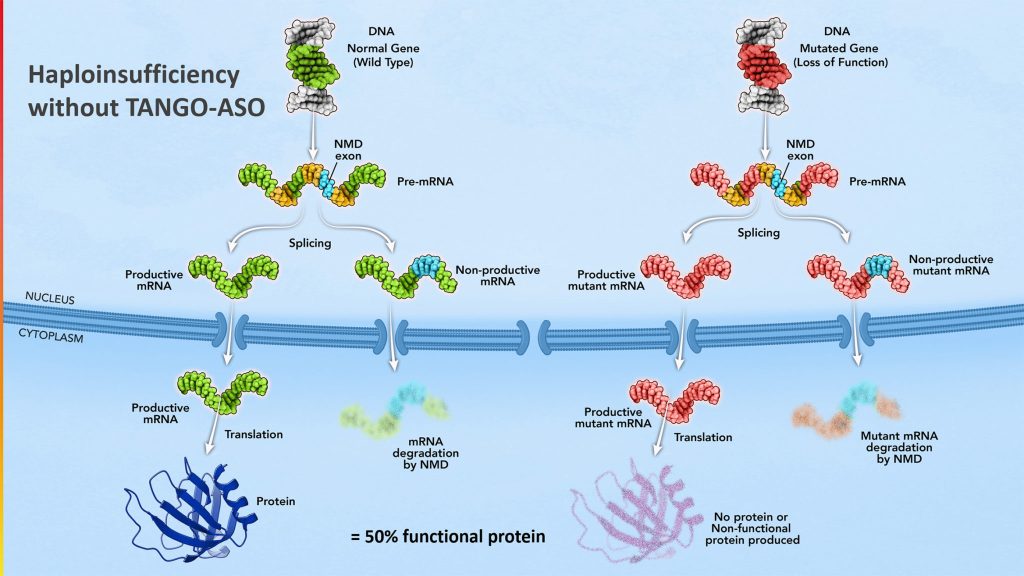
Stoke’s initial focus includes haploinsufficiencies and diseases of the central nervous system and the eye, although proof of concept for its proprietary approach has been demonstrated in other organs, tissues, and systems. Stoke’s first compound, STK-001, is in clinical testing for the treatment of Dravet syndrome, a severe and progressive genetic epilepsy. Dravet syndrome is one of many diseases caused by haploinsufficiency (in which a loss of ~50% normal protein levels causes disease).
Stoke is also pursuing the development of STK-002 to treat autosomal dominant optic atrophy (ADOA), the most common inherited optic nerve disorder. In August 2022, Stoke enrolled the first ADOA patient in a two-year prospective clinical study.
GEN Edge interviewed CEO and director Edward M. Kaye, MD, to learn how he was brought onto the leadership team at Stoke Therapeutics and the company’s latest progress treating rare genetic diseases with ASOs.
GEN Edge: Edward, what attracted you to be a part of Stoke Therapeutics?
Edward Kaye: I worked with Adrian on the Spinraza program when I was working with Genzyme. I got to know him as a scientist and respected his work. He called me and said we’ve got this new company interested in protein up-regulation using RNA splicing. I realized this is a fascinating approach to upregulating protein.
Most people have thought about upregulating proteins using messenger RNA (mRNA). There’s a lot of interest in mRNA because of vaccine development with Moderna. But one thing that is complicated with mRNA is that it breaks down immediately—all these endonucleases break it down. Getting it delivered into a cell to express a protein was challenging. It worked for things like vaccines because lymphocytes immediately took it up. However, other indications were a lot more challenging. Certainly, gene therapy is a way to upregulate a missing protein permanently. Still, the challenge is to deliver the exact amount of protein in the right cells using the current number of capsids and our gene therapy mechanisms.
This is a good approach to correct autosomal dominant diseases, where you’re missing 50% of the protein (haploinsufficiency). As a pediatric neurologist and biochemical geneticist, this was something that had stymied me. Most of the diseases we worked on were recessive diseases. At Genzyme, we avoided the dominant diseases! But now there’s an approach that we can titrate the exact amount of protein. We can take it from 50% to 100% and hopefully restore that protein and have a biological effect on the disease. It was an interesting opportunity to go after a set of previously difficult diseases.
I decided that this was too interesting of a scientific platform to miss out on, and I joined the company and came in as CEO in 2017. It’s been a fun experience going after new targets, especially since we are focused on rare diseases, most of which are pediatric. It was in my wheelhouse.
Kaye: I had taken care of patients with Dravet syndrome and realized this was a challenging disease. I experienced firsthand that it’s more than just a seizure disorder. Dravet syndrome is considered one of the more common genetic seizure disorders. But when you take care of these patients, there’s more than simply seizures. There are other aspects of the disease, such as behavioral problems: sleep, gait, and speech problems. These children reach a period of stagnation in their development; for the first couple of years of life, they seem to be developing quite typically, and then they stop progressing.
That was as upsetting to families as the seizures. The treatments are anti-epileptic, which is fine, and they treat seizures. But they need to address the other aspects. Treating genetic epilepsy by going after the primary genetic cause seemed to be a very reasonable approach. In other words, if you’re missing the sodium channel and could upregulate that sodium channel to get it back up to a hundred percent, it should significantly improve these patients. That was the hypothesis that we had developed. We’re in the clinic now, and we can demonstrate that that is the case.
GEN Edge: How does delivery go into the choice of drug modality for pediatric CNS diseases?
Kaye: One of the reasons why we decided to go after the CNS is that antisense oligonucleotides (ASOs) are indeed quite good, but they don’t get into every organ or every cell type. Adrian had shown with Spinraza that if you deliver ASOs by intrathecal delivery, you get nice biodistribution throughout the brain. So, that was already precedent with the use of Spinraza. We knew we could get into very diffuse parts of the brain by giving an intrathecal administration. It does not work with this particular ASO if you give it systemically because it doesn’t cross the blood-brain barrier. You could get nice delivery into the eye with an intravitreal injection. One of the reasons why we focused on the CNS and the eye was a delivery issue.
That doesn’t mean we wouldn’t go after other organs and diseases. But we would have to have a delivery mechanism that went into those organs and tissues. Stoke has a whole program where we look at different ASO-backbone chemistries for delivery. We’ve looked at conjugation with fatty acids or antibodies. We are exploring and looking at other tissues that have already been demonstrated. One is the kidney. The other tissue is the lung. Others have demonstrated that if you give ASOs by an aerosol delivery, it gets into many parts of the lung. We’ve thought about muscle and heart, but we have to have a different delivery system. Several companies we’ve talked to have systems that might be amenable to us.
We also look at the TANGO signature. Only some genes have the TANGO signature. Specific genes are easy to regulate where you have a lot of RNA messages that are non-productive that you can turn into productive. Each gene and each disease is different. We go through a rigorous process before we decide on a new therapeutic area or target we want to go after.
GEN Edge: How does Stoke Therapeutic manufacture ASOs?
Kaye: We use a couple of contract manufacturers and go through a fairly rigorous process to audit other companies. One nice feature about using ASOs is that there’s been a lot of work done on manufacturing. There are a lot of contract manufacturers that can make these, so we don’t have to manufacture them ourselves. It’s not that complex. It’s not like gene therapy, which is very complicated.
You have to deal with the percentage of empty versus full capsids. Many companies have said this is important, and we have to do it ourselves. But that’s a massive investment. I remember looking at gene therapy when I was at Genzyme. We figured out this’s a half-billion-dollar investment to invest in a manufacturing facility to do it right.
If you can contract out and have quality manufacturers know how to do it, why reinvent the wheel if you don’t have to while spending money. So, that’s a fortunate thing for us that we can use contract manufacturers to make our product. The cost of goods is certainly much less than what you’d have to spend on things like gene therapy, which are costly and difficult to make.
GEN Edge: What milestones does Stoke Therapeutics have in its sights?
Kaye: Dravet syndrome is a major focus right now. We hope to get data from our Phase II that will inform us of the trial design for Phase III. We want to demonstrate proof of concept in humans with Phase II and then go on to Phase III, a placebo control study for registration and commercialization. That’s step number one.
We also plan to be in the clinic with our next program next year. That would be for dominant optic atrophy, the leading genetic cause of optic atrophy. This is similar to Dravet in that it’s a nonsense mediate decay (NMD) exon. You’re missing 50% of the OPA1 protein —an essential protein for mitochondrial function and structure.
These children are born with normal vision. Then, somewhere at the end of the first decade of life, typically in school when they have a vision test, they find out that their vision is affected and can’t be corrected by glasses. It’s a non-refractive error. Then you get sent to an ophthalmologist or a neuro-ophthalmologist, and somebody picks up that there’s optic atrophy, which is just a whiteness or a power of the optic nerve. Because these retinal ganglion cells are very energy dependent, they slowly die. But it’s a very slowly progressive process that takes decades.
If somebody finds out they have optic atrophy and are unsure why, they can undergo standard testing. People are currently being tested and explicitly diagnosed with autosomal dominant optic atrophy. But again, we’ve demonstrated recently that you can get the ASOs into those retinal ganglion cells. More importantly, we’ve shown that we can regulate protein very nicely—and even more importantly, we can improve oxidative phosphorylation in these cells. It is a good demonstration that we can get the ASO where we want it to be, and it has an effect. We like that one and expect to be in the clinic next year with that program and move that along.
We also have a collaboration with Acadia. That’s a program where we have three targets that we’ve identified, two of which we’ve announced publicly. One is Rett syndrome—a fairly common progressive disease that includes not only epilepsy but also autistic spectrum and a neurodevelopment problem. The other is genetic epilepsy. But more importantly, it has a big neurodevelopmental component to it. With both or at least one of these programs, we’d like to address some of these progressive diseases’ neurologic problems.
Rett syndrome is very similar. There are affected girls, and there could also be more severely affected boys that appear normal. In the first few years of life, they start to have these atypical movements in this progressive autistic spectrum disorder. There’s certainly evidence in mouse models that you probably can arrest this progression again if you can replace that protein, MECP2, which is a critical protein for brain function. That would lead to [therapeutic programs for] not only epilepsy but nerve developmental disorders.
There’s a host of haploinsufficient diseases that affect cognition, so that could be a fascinating area for us too. Nobody has cracked that. No one has gone after these developmental disorders. Based on animal work, we now know that it’s not all prenatally determined that this disorder continues to progress. Especially something like Rett syndrome, where these girls are quite normal at birth and then seem quite normal for four to six years, and then suddenly have this deterioration. So if you can identify these early, I think we could also affect some of these symptoms.
This has been a challenging time for small- and mid-cap companies, but we saw this coming two years ago, I thought it would happen a year earlier, but we went public, did financing, and used our at-the-market (ATM) offering. Part of it was to develop a war chest to survive the downturn. We have enough cash to get us into 2025, which is a nice position. We’re not worrying about running out of money. We’re just focused on trying to ensure we do the best science.

