December 1, 2017 (Vol. 37, No. 21)
Experts in the Field Speak to the Ways CRISPR Is Used
Genome-Wide CRISPR Screen Identifies New Class of Regulatory Proteins
Using a CRISPR-based genetic screen, Cellecta recently identified, for a client, a class of proteins responsible for regulating gene expression of a genetic element present in the 5′-untranslated region (5′-UTR) of a gene involved in a specific disease indication.
For this study, we introduced Cellecta’s 80K Genome-Wide CRISPR Knockout Library, which expresses CRISPR single guide RNAs (sgRNAs) targeting all known human protein-coding genes (four sgRNAs/gene), into two isogenic cell lines. Both cell lines were engineered with a green fluorescent protein (GFP) expressed from the same promoter.
However, one cell line had the genetic regulatory element of interest inserted between the promoter and GFP reporter, and the other did not. At a few different time points, then, both cell lines were sorted by flow cytometry to separate the cells that brightly fluoresced from those with low fluorescence (Figure).
 |
|
Donato Tedesco, Ph.D., Director of R&D, and Paul Diehl, Ph.D., COO, Cellecta
|
The relative abundance of cells transduced with each sgRNA in the populations of high- vs. low-fluorescent cells collected at each time point was assessed by high-throughput sequencing. This analysis revealed a number of sgRNAs overrepresented in the low-fluorescing population that had the genetic element of interest, as opposed to the control cells that did not. These sgRNAs targeted a small group of genes that appear to regulate expression of this element since their disruption reduced expression of the GFP reporter.
In conclusion, with this one assay we were able to screen all human protein-coding genes and identify a set of proteins responsible for controlling this therapeutically interesting genetic regulator.
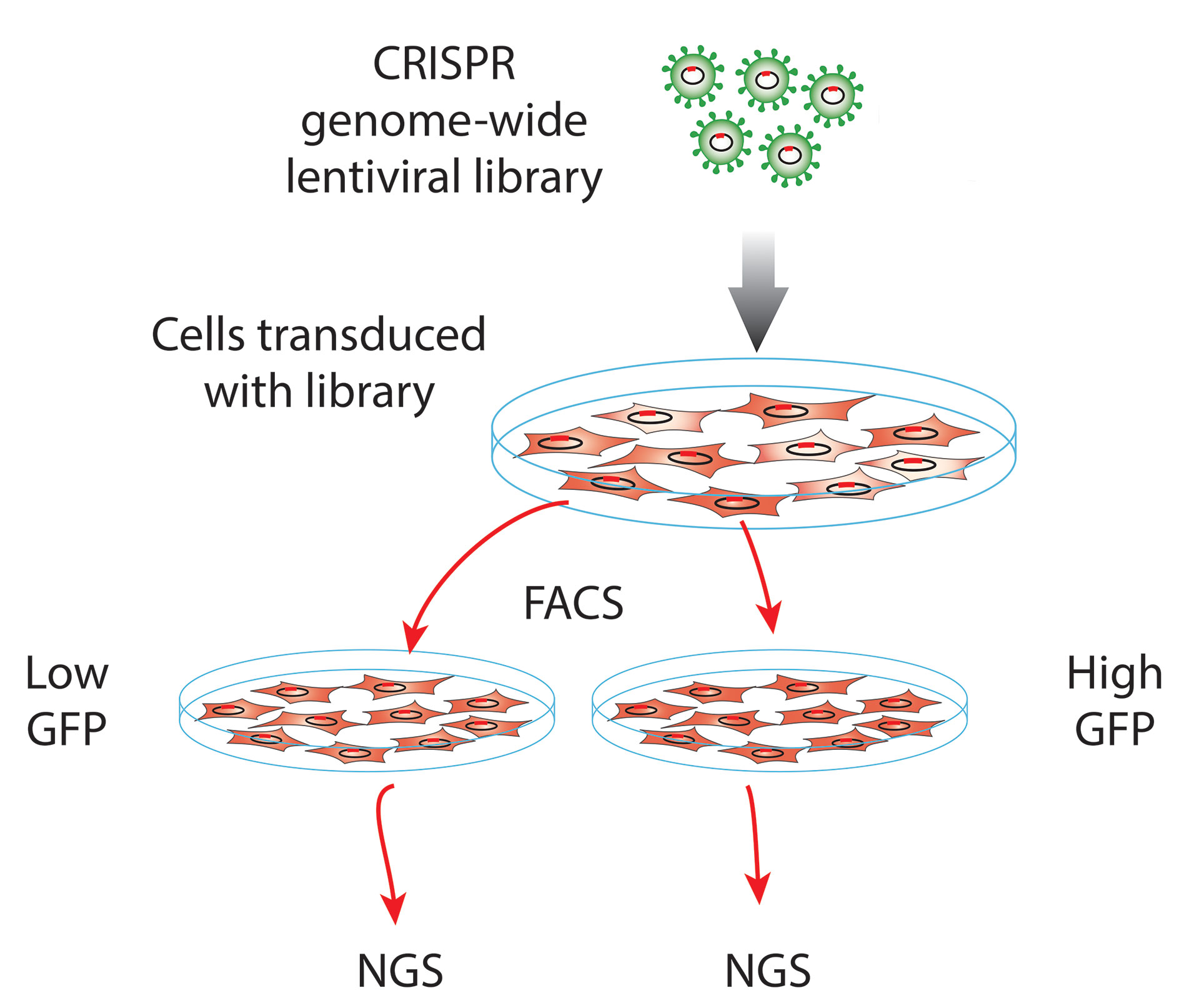
Figure. Cellecta’s CRISPR technology enables genetic screens and the development of genome-edited cell lines.
CRISPR/Cas9 Arrayed Screening with Synthetic crRNA in Primary T Cells as a Platform to Accelerate HIV Therapy Development
For years, high-throughput arrayed functional genomics screening has been a standard for understanding biological pathways. The ability to analyze phenotypes on a one-well-per-gene basis supports sophisticated phenotypes, including high-content assays, and enables a richer investigation of biological mechanisms than a pooled screen. There are hundreds of examples of these loss-of-function screens with RNAi, but only recently have we seen arrayed studies with CRISPR/Cas9-mediated gene knockout.
A 2016 publication (J.F. Hultquist et al., “A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells,” Cell Rep. 17(5), 1438–1452 [2016]) elegantly demonstrates the power of arrayed CRISPR screening using the Dharmacon Edit-R™ synthetic guide RNA platform. Their screen leveraged a robust, high-throughput assay system that includes electroporation of CRISPR/Cas9 ribonucleoproteins (RNPs) consisting of Cas9 protein with a synthetic CRISPR RNA (crRNA) and tracrRNA guide RNA.
The study was focused on identifying novel targets that impact HIV infectivity. As a proof of concept, the researchers performed a high-throughput CRISPR screen of 146 different RNP complexes targeting 45 genes involved in HIV integrase function. In a 96-well format, RNPs were electroporated into primary T cells isolated from several different blood donor samples.
The robustness of this approach was validated by measurement of phenotype, gene-editing percentage, and gene knockout. The authors were able to confirm expected phenotypes when knocking out several host factors and also discovered new gene targets. Thus, they demonstrated the usefulness of a scalable, arrayed CRISPR knockout screening approach to identify novel host factors for HIV pathogenesis and infection. This platform is expected to have utility in elucidating other biological pathways in primary T cells as well as other primary cells important in human disease.

Louise Baskin, Senior Product Manager, Dharmacon, a Horizon Discovery Group Company
Overcoming Bottlenecks in Screening for Clones
As use of the CRISPR/Cas systems expands, new bottlenecks have emerged. A significant bottleneck is the screening for clones containing induced mutations and the determination of the zygosity. While sequencing can be used as a screening method, cost and time requirements prevent its widespread adoption. As a result, scientists are turning to low-cost screening steps to reduce the number of individuals and pools sequenced.
Heteroduplex cleavage assays are an efficient method to screen for CRISPR-induced modifications, while simultaneously allowing the determination of gene editing frequency. Heating, then slowly cooling PCR products allows the formation of heteroduplexes between wild-type and edited amplicons.
After heteroduplex formation, the sample is treated with an endonuclease, such as T7 Endonuclease I, which recognizes and cleaves at DNA mismatches.
Advanced Analytical Technologies, Inc. (AATI) has developed a robust T7 endonuclease assay (AccuCleave T7CE) that seamlessly integrates with the Fragment Analyzer™ Automated CE System, allowing for accurate determination of CRISPR gene editing frequencies. To determine the zygosity, AATI developed statistical models based on cleavage ratios that allow researchers to characterize mutations made in monoclonal diploid cell lines or organisms. AATI has fully automated the CRISPR detection and quantification workflow, enabling rapid screening of edited cell lines, animals, and plants—putting crucial results in researchers’ hands sooner.

Kyle Luttgeharm, Ph.D., Application Scientist, AATI
Alt-R S.p. HiFi Cas9 Nuclease 3NLS: Highly Specific Genome Editing, Even under Challenging Conditions
An ongoing concern for many researchers is potential off-target effect introduced by the CRISPR/Cas9 system. Existing CRISPR guide RNA design tools often fail to effectively predict off-target sites. Delivery of the genome-editing reagents as an RNP complex, reduces but does not eliminate off-target editing. Some popular Cas9 mutants that provide improved specificity compromise on-target efficiency, resulting in poor utility when delivered in RNP format.
The latest addition to the highly potent Alt-R CRISPR-Cas9 system—Alt-R Streptococcus pyogenes HiFi Cas9 Nuclease 3NLS—achieves a perfect balance between genome editing efficiency and accuracy. IDT scientists took a unique approach to evolve the Cas9 nuclease into a more precise genome-editing tool.
By screening over 250,000 mutants, they discovered this enzyme effectively eliminates the risk of unpredictable off-target cleavage typically observed with the use of wild-type Cas9. Unlike rationally designed Cas9 variants that suffer from a moderate to severe loss of on-target activity, the Alt-R S.p. HiFi Cas9 Nuclease 3NLS preserves a high level of editing efficiency at intended on-target sites while radically reducing off-target effects.
Its superior performance has been validated by a number of prominent laboratories conducting translational research with various biological systems. The release of this high-fidelity Cas9 enzyme is a significant step toward the therapeutic use of CRISPR.

Brian Wang, Ph.D., Genomic Tools Market Development Manager, Integrated DNA Technologies
Type 1 Diabetes Mouse Model
The Jackson Laboratory’s Model Generation Services (MGS) team has extensive experience using CRISPR technology to create genetically modified mice, not just in the C57BL/6J strain, but in a wide variety of inbred mouse strains. This capability is significant because when developing a new genetic model, consideration of background genetic effects is essential to the therapeutic relevance of the new strain.
In one example, David Serreze, Ph.D., a principal investigator at The Jackson Laboratory, was interested in testing the hypothesis that the Aicda gene, which is required for class switch recombination/somatic hypermutation in B lymphocytes, is important in the development of type 1 diabetes. He selected the NOD/ShiLtDvs inbred strain, a polygenic model for autoimmune type 1 diabetes.
Creating the new strain directly on the NOD background avoided time consuming and expensive backcrossing, while also significantly reducing the number of mice needed.
The CRISPR-generated NOD.Aicda−/− mice were compared with the parental NOD strain. Type 1 diabetes development was decreased in the NOD.Aicda−/− mice, suggesting that class switching is required for the development of type 1 diabetes.
Aicda is known to be a part of the same functional pathway as RAD51. NOD mice were dosed with the small molecule 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid, which inhibits RAD51-mediated homologous recombination. Similar to NOD.Aicda−/− mice, type 1 diabetes development was significantly decreased in the treated mice.
The creation of CRISPR/Cas9-driven modifications of the Aidca gene on the NOD background strain by The Jackson Laboratory’s MGS team showed that the Aidca/RAD51 functional pathway might be a target for developing compounds that can block type 1 diabetes development. This work is potentially translatable to the clinic as an approach to block the development of type 1 diabetes.

David S. Grass, Ph.D., Senior Director, Genetic Engineering, Transgenic Genotyping, and Reproductive Sciences, The Jackson Laboratory
Transfection Technology Aids Development of CRISPR-Based Therapies for Multiple Monogenic Diseases
CRISPR is a transformative technology at the crossroads of basic research, drug discovery, and human therapeutics. Whether for use in basic research or as a therapeutic agent, CRISPR gene-editing machinery must be delivered to the relevant cell populations. As research grows more sophisticated and studies move to the clinic, the delivery of CRISPR machinery—whether in the form of proteins, RNA, or DNA—becomes challenging as the target cells are frequently more difficult-to-transfect cells, such as primary cells or induced pluripotent stem cells (iPSCs).
MaxCyte offers an easy-to-use, highly flexible delivery platform that is based on Flow Electroporation™ technology and that facilitates efficient gene editing. MaxCyte’s platform is designed for high performance, reproducibility, and scalability, and it can provide a regulatory pathway to support gene-editing applications ranging from R&D to global gene therapy.
In 2014, MaxCyte joined collaborative research efforts to develop potentially curative CRISPR-based therapies for multiple monogenic diseases such as X-linked chronic granulomatous disease (X-CGD) and sickle-cell disease (SCD). MaxCyte’s partners in these efforts include Harry L. Malech, M.D., chief of the Genetic Immunotherapy Section at the National Institute of Allergy and Disease, and John F. Tisdale, M.D., senior investigator in the Molecular and Clinical Hematology Section at the National Heart, Lung, and Blood Institute.
Cells harvested from X-CGD or SCD patients were edited ex vivo using a CRISPR-mediated nuclease system and MaxCyte’s delivery platform. High transfection efficiencies and cell viability of patient cells provided for clinically relevant gene correction rates for both indications.
In addition, short- and long-term in vivo engraftment models demonstrated the restoration of function to immune cells affected by X-CGD. (S.S. De Ravin et al., CRISPR/Cas9 Gene Repair of Hematopoietic Stem Cells from Patients with X-Linked Chronic Granulomatous Disease,” Sci. Transl. Med. 9(372), (2017); S.S. De Ravin SS et al. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat Biotechnol. 2016, 34(4): 424–9.)
Both collaborations remain in progress and are focused on bringing these treatments to market and making curative gene editing therapies a reality.

Linhong Li, Ph.D., Director of Cell Engineering, MaxCyte
Expedite the Analysis of CRISPR/Cas9 Products by Automating the Measurement of Editing Efficiency
Using CRISPR/Cas9 to make precise gene modifications requires robust analytical tools to verify the successful generation of on-target mutations, and to measure the efficiency of the experiment during protocol optimization. Traditional CRISPR/Cas9 analysis is bottlenecked by the lack of an efficient analytical tool to confirm editing events in a large pool of samples.
Classical analysis protocols are time consuming and not amenable to high-throughput analysis. PerkinElmer’s LabChip® GX Touch™ Nucleic Acid Analyzer uses microfluidic electrophoresis to automate the qualitative and quantitative analysis of CRISPR/Cas9 modified products. This reduces the number of samples that need to be confirmed by sequencing and increases the capacity of the CRISPR workflow.
The LabChip GX Touch Nucleic Acid Analyzer offers real-time analysis of the presence of desired mutations in less than 60 seconds per sample. The mutations are identified by electrophoretic mobility shifts of the heteroduplex strand, a signature peak of on-target gene editing. In combination with digestion of the heteroduplex strand with T7 Endonuclease I and analysis of the resulting fragments, the on-target efficiency of gene editing can be determined.
The LabChip GX Touch Nucleic Acid Analyzer allows a pool of samples to be screened in a high-throughput fashion. The editing efficiency of up to 384 samples can be analyzed within a few hours, resulting in a streamlined protocol for the analysis of CRISPR/Cas9 products.
For research use only. Not for diagnostic procedures.

Zhiyong Peng, Ph.D., Application Scientist, Applied Genomics, PerkinElmer
Proxy-CRISPR Experimentation
MilliporeSigma has a highly active R&D program to explore CRISPR function and practical utility for genome editing. MilliporeSigma, like others, has found that many natural bacterial CRISPR systems cannot function efficiently in human cells to support practical genome editing work. The Cas9 protein from Streptococcus pyogenes (SpCas9), however, works surprisingly well in the chromatinized context of the human genome.
Therefore, SpCas9 has become widely adopted, and MilliporeSigma chose it to assume the role of “hypothesized chromatin disruptor” in its initial proxy-CRISPR experiments.
MilliporeSigma made a mutant of SpCas9 that lacks DNA cutting activity (dead-SpCas9 or dSpCas9) and targeted it to adjacent locations of less active CRISPR systems (such as FnCpf1, FnCas9, CjCas9, etc.). To the company’s surprise, proximal targeting of dSpCas9 had a strong and consistent positive effect on these various bacterial CRISPR systems in human cells.
Proxy-CRISPR can enhance genome editing projects in at least three important ways: (1) it can boost genome editing activity, including both gene knockout and knockin; (2) it can enable targeting of DNA cutting closer to desired mutation sites, which exponentially increases knockin activity; and (3) it creates a requirement for two binding events to achieve knockout/knockin, thus reducing the frequency of off-target cutting.

Martha S. Rook, Ph.D., Head of Gene Editing and Novel Modalities, MilliporeSigma
Enabling Protein Biology Studies in a More Natural Biologic Context
Tag sequences are often incorporated into expressed proteins facilitating characterization of various aspects of protein function. However, traditional overexpression models can result in experimental artifacts by removing proteins from the native genomic context and changing stoichiometry with interacting partners.
Endogenous gene tagging with CRISPR allows protein studies under physiologically relevant conditions. However, the large size of many tags results in low insertion efficiency, and detection is often restricted to antibody-based methods that limit throughput, sensitivity, and the ability to obtain quantifiable results.
Promega scientists have overcome these limitations by combining an 11-amino-acid peptide tag with sensitive bioluminescent detection, HiBiT, and CRISPR editing. We find efficient insertion of the HiBiT tag by using a single-stranded oligodeoxynucleotide donor molecule, which is ordered directly, eliminating the need for molecular cloning.
HiBiT is detected using a simple add-and-read bioluminescent method that has over 7 logs of linear dynamic range. Such sensitivity enables quantification of HiBiT-tagged proteins in pools of edited cells, rendering the process from editing to assay as quick as 24–48 hours.
We have used endogenous HiBiT tagging to study the regulated abundance of proteins HIF1A and BRD4 and found that expression from the endogenous locus results in significantly improved assay response compared to transient overexpression. Our work has demonstrated the HiBiT tag to be highly amenable to CRISPR knockin, enabling protein biology studies in a more natural biologic context.

Amy Landreman, Ph.D., Global Product Manager, Promega
A CRISPR Way of Modifying Animals
CRISPR/Cas9 has transformed the generation of modified animals by enabling gene modifications in a multitude of previously intractable species. The CRISPR toolbox has itself evolved rapidly with novel synthetic technologies that have further accelerated and increased the probability of project success.
High-efficiency CRISPR reagents such as chemically modified synthetic single-guide RNA (sgRNA) from Synthego have set a new high bar for successful genome editing. Data from the laboratory of Joseph M. Miano, Ph.D., associate director of the Aab Cardiovacular Institute at the University of Rochester Medical Center, demonstrates that delivering sgRNA and Cas9 protein (as ribonucleoproteins or RNPs) to the one-cell animal embryo via microinjection has resulted in the rapid production of new genetic animal models that previously would be considered risky or even challenging to engineer.
This optimized approach to generating and genotyping “CRISPRized” animals has led to a nearly 100% success rate of germline transmission in the animal models, enabling rapid generation of genetically modified animals in the span of weeks with almost a guarantee of success. This level of success has led Dr. Miano to comment on how well Synthego’s sgRNAs work the mouse zygote: “Not only has Synthego saved us valuable time, we are getting higher efficiency precision editing by HDR. We have completely switched over to Synthego’s sgRNA and Cas9 protein for RNP injections, and we are not looking back.”
![]()
Abhi Saharia, Ph.D., Director of Product Management, Synthego
For research use only. Not for diagnostic procedures.

