
Mary F. Lopez, Ph.D.
Learn how a proteogenomics approach can improve your investigational power.
Combining genomic and proteomic techniques can reveal important insights to unlock complex biological function. The exploration of methods for integrating large genomic and proteomic data sets is gaining traction due to improved bioinformatics and the public availability of well-organized, searchable data. Also, by merging RNA sequencing with mass spectrometry and improving bioinformatics, scientists can strategically select from a much broader range of investigational techniques and take advantage of complementary information. When one avenue of investigation closes, another may offer a new route to discovery—either building on previously inconclusive results or offering altogether new insights. Multiple studies in the past several years have demonstrated that transcriptional profiling data do not necessarily correlate with protein expression data, confirming that the two types of information are not duplicates but, instead, may be synergistic in terms of broadening our understanding of basic biology.1
In this brief overview, three recent examples illustrate how a “proteogenomics” approach can improve investigational power by enabling a progression of approaches that lead to actionable conclusions. When it comes to translational medicine, proteogenomics may provide better prospects for revealing biomarkers, assessing disease states, and identifying the complex mechanisms behind biological function.
Histone Depletion in Cellular Aging
In partnership with Victoria Lunyak, Ph.D., at the Buck Institute for Research in Aging, our team at BRIMS worked to combine transcriptomics with proteomics to investigate the expression of histone variant H2A in human fibroblasts.2 Previous research had pointed to histone depletion as causal in the incorrect chromatin binding that leads to cellular aging. By exposing one group of cells to a genotoxin, we were able to compare the histone expression of normal cells to those under genotoxic stress.
First, using Western blot analysis, we discovered that peptide sequences associated with H2A in the treated cells were lower than those in normal cells, as we had expected. Then we used qPCR to investigate whether or not the genes associated with H2A production were affected by the genotoxin. This revealed that, in the cells under stress, the genes associated with H2A production were highly expressed. Next, to confirm the Western blot findings, we turned to mass spectrometry to quantify H2A in treated and untreated cells. In this study, we developed a multiplexed SRM assay for quantitative measurement of histone H2A family proteins and, specifically, for histone H2A.X and phosphorylated histone H2A.X (γH2A.X) (Figure 1). This histone variant undergoes rapid phosphorylation of serine at position 139 by members of the PI3-kinase-like family in response to double-stranded DNA breaks.
Our investigations confirmed that chronic DNA damage impacts the expression of H2A family histones and leads to their depletion in senescent cells. Through the use of this integrated approach combining genomics with proteomics, we proposed that changes in histone biosynthesis and chromatin assembly may directly contribute to cellular aging.
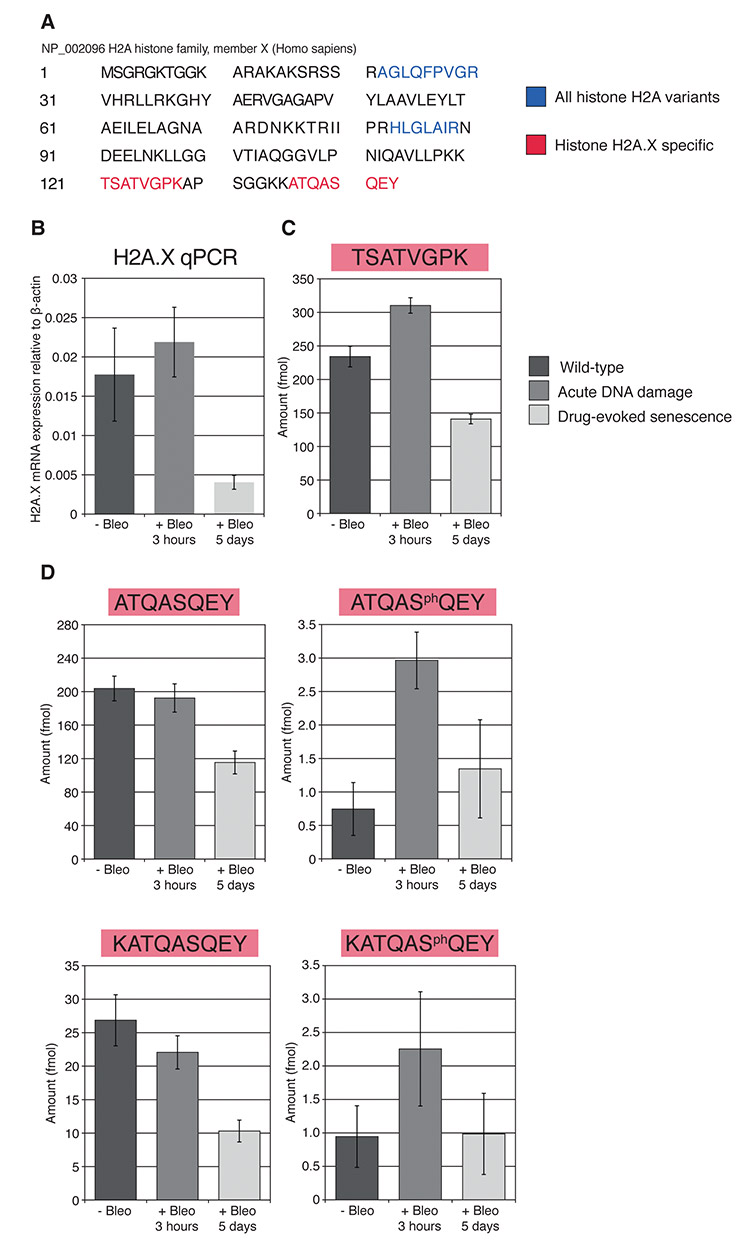
Figure 1. From Lopez, M.F., et al. (2012) Depletion of nuclear histone H2A variants is associated with chronic DNA damage signaling upon drug-evoked senescence of human somatic cells. Aging 4(11), 823–842.
ER-Positive Breast Cancer Biomarkers
Cancer researchers, in particular, are making strides via proteogenomics in their quest to identify which subtypes of cancer will respond to which potential treatments. As well, these researchers are combining data and techniques from both fields in creative ways to identify prognostic biomarkers for early disease detection.
Pavlou et al. integrated the transcriptome and proteome in a search for biomarkers to predict risk for recurrence in breast cancer patients with estrogen receptor (ER)-positive tumors.3 They meta-analyzed the public microarray data for survival-associated genes (89), comparing these with the breast cancer proteome data from a previous study. This effort produced a list of potential protein biomarkers.
The team developed a method based on mass spectrometry to identify proteins associated with ER-negative and ER-positive samples. Two proteins previously identified by the team, PTX3 (ER-negative) and ABAT (ER-positive), emerged as significantly linked.
Finally, the team analyzed results separately based on ER status. As expected, there were no notable differential expressions among protein levels for ER-negative samples; however, two proteins—KPNA2 and CDK1—were expressed by approximately twofold in ER-positive patients with poor prognosis. KPNA2 has been previously linked to cancer, including breast cancer, but this was the first observation of its prognostic potential for early-stage breast cancer in ER-positive tumors.
New Subtypes in Colorectal Cancer
Rather than treating and classifying tumors solely by the primary tumor's site of origin, researchers can now identify cancer subtypes based on their combined genetics and proteomics. Subtler subtyping may result in better treatments based on greater insight into protein targets.
Zhang et al. analyzed 95 colon and rectal tumor samples listed in The Cancer Genome Atlas (TCGA) and, using shotgun proteomics, identified 124,823 distinct peptides corresponding to 6,299,756 spectra in an assembly of 7,526 protein groups.4 They also identified 796 single amino-acid variants (SAAVs), corresponding to 64 somatic tumor variants, which they then validated using parallel reaction monitoring (PRM). Among these were 162 previously unreported SAAVs—64 of which matched RNA-seq data and corresponded to somatic variants from TCGA. Of the remaining SAAVs, 526 were listed in the Single Nucleotide Polymorphism database and are likely germline variants. Notably, the somatic variants were enriched in hypermutated samples and exerted a significantly stronger negative impact on protein abundance in the colon and rectal tumor samples.
The research team then examined the relationship between protein levels and mRNA copy numbers, along with the subsequent biological function of the gene product using KEGG. They found only a modest correlation between mRNA and protein levels. They did, however, find a strong cis-effect of copy number alterations (CNAs) on mRNA abundance.
The team separated genes with known CNAs, mRNA, and protein measurements into groups according to focal amplification regions, focal deletion regions, and nonfocal regions, finding that focal amplifications had the strongest cis-effects on both mRNA and protein abundance. Chromosomes 20q, 18, 16, 13, and 7 contained the five strongest hot spots driving global mRNA abundance variation. The 20q amplification was associated with the largest global changes in both mRNA and protein levels. One of the 79 genes in the 20q region, HNF4A, is a TCGA-identified transcription factor implicated in colon and rectal cancers.
With low correlation between mRNA and protein levels, the researchers subtyped the colon and rectal cancer samples according to proteomics data, identifying five subtypes. HNF4A abundance was significantly higher in subtype E tumors, as compared to normal colon samples. This proteogenomic approach revealed the importance of chromosome 20q amplification in colon and rectal cancers and the subsequent role of HNF4A.
Conclusions and Challenges
These cases highlight the importance of innovation in investigational methods. Researchers can now move from dataset to dataset, employing vastly different technologies—both in parallel and sequentially—to drill down into highly complex systems. But even though proteogenomics holds much promise, challenges remain.
To leverage the technologies and the huge accumulated knowledge from proteomics and genomics, research teams must conscript from multiple disciplines. Although this is not an entirely new concept, investigators with the breadth of knowledge to oversee proteogenomics projects are the exception, not the norm. In this regard, collaboration across multiple principal investigators is paramount to success.
Furthermore, by adding petabytes of proteomics data to terabytes of genomics data, the complexity of bioinformatics increases significantly. If there were ever a reason to adopt “big data” techniques and develop new statistical methods for biological investigation, proteogenomics renders this even more imperative. This creates an urgent need for purpose-built software for translational applications that can handle quantitative data from hundreds of samples and integrate the results with biological pathways. To date there is a dearth of these types of analysis platforms, especially in the proteomics area, which has typically lagged behind genomics.
Studies utilizing multiple techniques to unlock biological function will take longer and require the aggregation of financial and human resources across disciplines. The depth of insight that a proteogenomic investigational approach affords, however, has the power to move translational medicine far closer to the realm of possibility.
Mary F. Lopez, Ph.D. ([email protected]), is director of the Thermo Fisher Scientific Biomarker Research Initiatives in Mass Spectrometry (BRIMS) Center.
References:
1 Vogel, C., et al. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature Reviews Genetics 13(4), 227–232. doi: 10.1038/nrg3185
2 Lopez, M. F., et al. (2012). Depletion of nuclear histone H2A variants is associated with chronic DNA damage signaling upon drug-evoked senescence of human somatic cells. Aging 4(11), 823–842.
3 Pavlou, M. P., et al. (2014). Integrating meta-analysis of microarray data and targeted proteomics for biomarker identification: Application in breast cancer. Journal of Proteome Research 13, 2897–2909. dx.doi.org/10.1021/pr500352e
4 Zhang, B., et al. (2014). Proteogenomic characterisation of human colon and rectal cancer. Nature. doi: 10.1038/nature13438

