

In Part I of this article, which appeared in the January 2022 issue of GEN, William A. Haseltine, PhD, discussed findings that can help us understand why some people resist COVID-19 more effectively than others. He argued that we can use these findings to help us shore up the body’s defenses against SARS-CoV-2. Here, in Part II, Haseltine discusses biochemical details of the immune system’s struggle with the virus, and how they may inform the development of antiviral drugs.
The extensive measures SARS-CoV-2 deploys to counter innate immunity are worth detailed study. They are attractive targets for antiviral drugs. Inhibit one or more of these proteins, and we tip the balance of innate immune suppression in our favor, limiting prolific replication early on and thereby reducing transmission. Drugs that inhibit viral countermeasures may also help those with limited innate immune function, including the elderly, control infection early on.
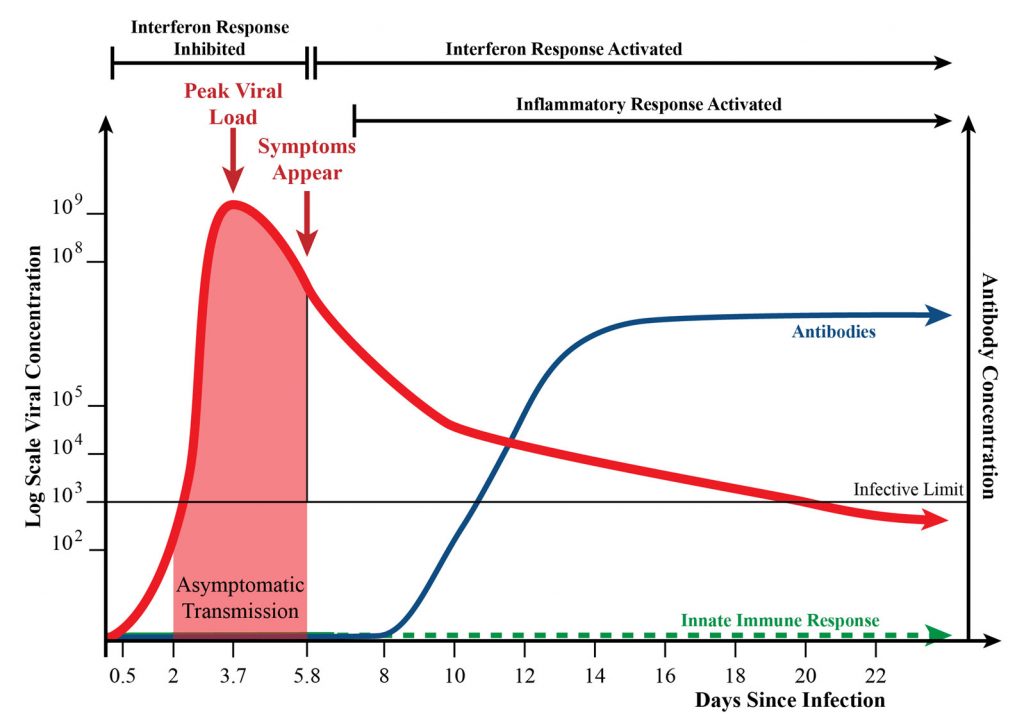
These possibilities were introduced in the prequel to the current article. The prequel, or “Part I,” appeared in the January 2022 issue of GEN, and it described phenomena such as the rise and fall in viral load and the start of antibody production in COVID-19 (Figure 1), and it detailed the role of interferon in the innate immune response to SARS-CoV-2 (Figure 2).
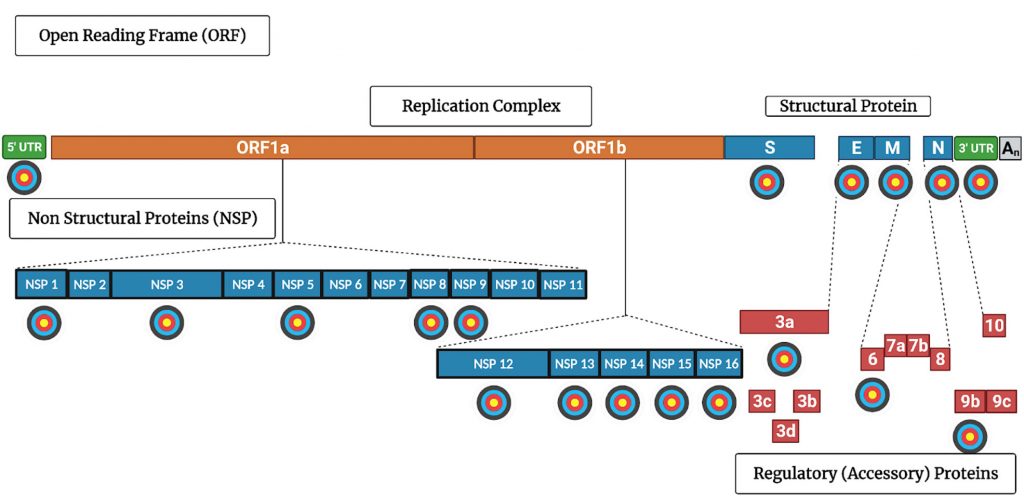
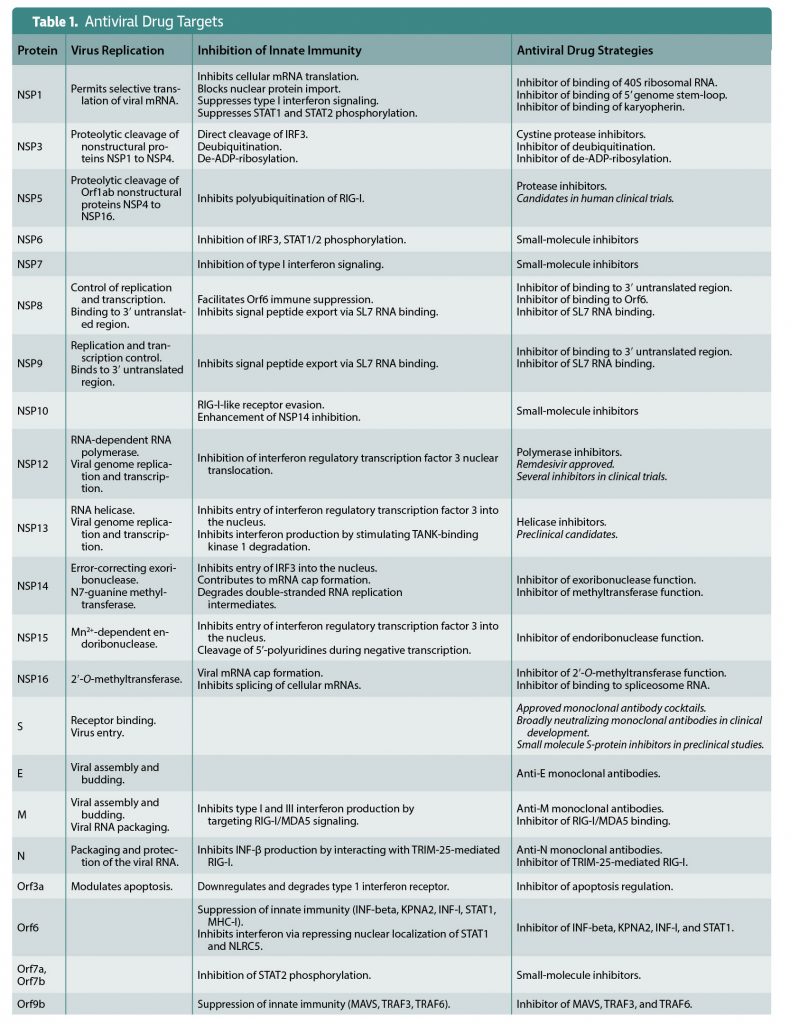
Viral countermeasures are explored further in this article, which serves as a sequel. Here, in “Part II,” the viral proteins that account for SARS-CoV-2’s countermeasures are discussed. More to the point, promising targets for antiviral drugs are indicated in Figure 3 and Table 1.
Over the past year and a half, studies from scientists around the world have unveiled many of SARS-CoV-2’s secrets. I am continually amazed as new discoveries appear almost monthly revealing an unprecedented variety of countermeasures that the virus deploys to ensure its survival. Some SARS countermeasures are well known from the study of other viruses. Many are new to science.
Concealment
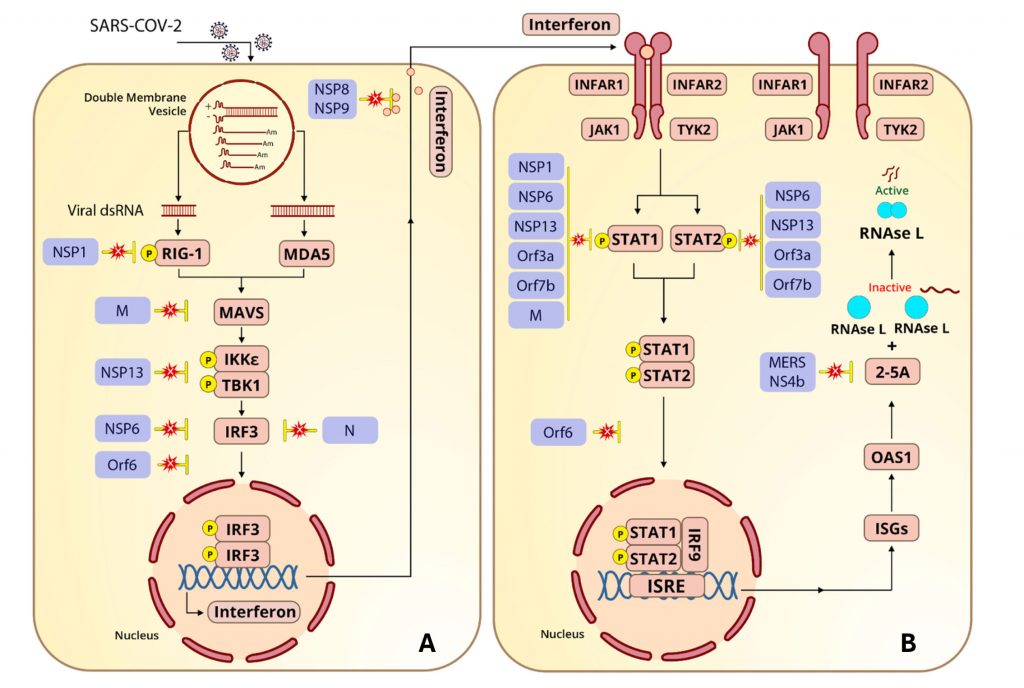
One of the first steps in virus replication is the formation of a double-membrane vesicle that serves as a privileged replication compartment. Viral proteins made early after infection reprogram the endoplasmic reticulum to create vesicles that wall off the viral genome’s replication and transcription machinery from the rest of the cell. The so-called replication-transcription double-membrane vesicle shields double-stranded RNA replication intermediates from retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) (Figure 2A).
The ability to breach the protective wall is the key 2′-5′-oligoadenylate synthetase 1 (OAS1) 46K protection. OAS1 46K penetrates the SARS-CoV-2 protective shield, whereas the 42K version does not. Recent experiments find OAS1 is tightly bound to the replication complex, most likely within the double-membrane vesicle. The entry pass into the privileged space is the lipid covalently coupled to the carboxy terminus of OAS1 46K.
The 46K protein has the lipid, and the 42K version does not. Disruption of replication is so extensive that the double-membrane vesicles themselves fragment. The 46K OAS1 inhibition by OAS1 46K is autocatalytic. The release of partially digested RNA from the disrupted replication compartment stimulates RIG-I and MDA5, leading to the production of additional interferon-stimulated genes, including OAS1 itself.
The formation of a double-membrane vesicle is a common property of many RNA viruses that replicate in the cytoplasm. These RNA viruses including all coronaviruses, flaviviruses (dengue and West Nile virus), and picornaviruses. The rs10774671 (G) allele that encodes the OAS1 46K inhibits replication of many of these viruses as well. Two exceptions are worth mentioning. Both SARS-CoV-1 and MERS are far more deadly than SARS-CoV-2. Of those infected with MERS, 30% die. SARS is 10% fatal. Both of these viruses are resistant to the OAS1-RNAse L. MERS encodes a protein that degrades 2′-5′-oligoadenylate, short-circuiting this pathway. How SARS-CoV-1 evades RNAse L remains a mystery. We can only hope that SARS-CoV-2 does not acquire similar resistance.
Camouflage
Viral messenger RNAs must leave the replication transcription compartment to encounter the protein synthesis machinery. They exit via a one-way pore in the double-membrane vesicle. Once in the cytoplasm, another series of innate immune alarms lie in wait to detect foreign RNAs. Upon synthesis in the nucleus, cellular messenger RNAs are modified by a set of enzymes that add a methylated and a 3′ polyadenylated tail. These enzymes are not present in the cytoplasm. Not to be outdone, coronaviruses, including SARS-CoV-2, encode all the enzymes needed to produce a 5′ methylated cap and equip the viral mRNAs with a suitable 3′ polyadenylated tail.
Inhibiting interferon induction
Interferon and interferon-stimulated genes are the bêtes noires of SARS-CoV-2. So much so that deficiencies in interferon production and action are a major cause of serious disease and death following SARS-CoV-2 infection. Patients who have failed to make interferon or who have antibodies that neutralize interferon are overrepresented in hospitalized patients in need of intensive care, as are people who inherit deficiencies in many of the signaling proteins outlined in Figure 2. Therefore, it should come as no surprise that SARS-CoV-2 encodes numerous proteins that interfere with interferon synthesis.
Two viral proteins block the very first step in interferon induction, RIG-I and MDA5 activation. The viral nonstructural protein 1 (NSP-1) prevents phosphorylation of RIG-I. The viral nuclear protein (N) does double duty by blocking MDA5 activation, as does the viral membrane (M) protein as a direct inhibitor of mitochondrial antiviral-signaling protein (MAVS). The viral protein NSP3 inhibits the next step of interferon activation, MAVS-induced phosphorylation of IkB kinase ε (IKKε) and TANK-binding
kinase 1 (TBK1).
Three viral proteins team up to prevent the viral final step in interferon induction, phosphorylation, and translocation of interferon regulatory factor 3 (IRF3). NSP6 inhibits phosphorylation of IFR3, Orf6 inhibits its translocation to the nucleus, and the NSP3 papain-like protease degrades IFR3.
In addition to inhibiting interferon induction via IRF3, Orf6 also represses expression of major histocompatibility complex class I (MHC-I), a class of molecules that plays a critical role in the production of type II interferons. Orf6 suppresses MHC-I by inhibiting nuclear localization of STAT1 (signal transducer and activator of transcription 1), which is required for interferon type I and III messenger RNA synthesis, and NLRC5 (NOD-like receptor family CARD domain containing five), a key transcription regulator for MHC-I.
As a final step, SARS CoV-2 inhibits the activity of any interferon that might slip through the induction blockade. The viral proteins NSP8 and NSP9 together bind the small SL7 RNA of the signal peptide export complex, preventing exit from the cell of any protein with a signal protein export sequence, including interferon. In doing so, SARS-CoV-2 compromises a key interferon function, alerting nearby cells to impending danger and initiating the adaptive immune response.
Inhibiting production of interferon-stimulated genes
SARS-CoV-2 encodes a suite of proteins to inhibit the induction of interferon-stimulated genes, including OAS1. We have seen how interferon binding to the interferon-α/β receptor (IFNAR) stimulates the JAK-STAT pathway. The viral proteins NSP1, NSP6, and NSP13; the viral membrane protein; and two of the viral accessory proteins, Orf3a and Orf7a, work in concert to inhibit the phosphorylation of STAT1 and STAT2. Orf6 does double duty, blocking STAT1/2 and the INFR3 complex from entering the nucleus.
Inhibition of synthesis of all cellular proteins
Upon infection, SARS-CoV-2 silences the production of any antiviral protein that may be made in reaction to viral infection including OAS1 and other interferon-stimulated genes.
The first viral protein made upon infection, NSP1, blocks translation of all cellular messenger RNAs while permitting synthesis of viral proteins. NSP1 has two activities that make it capable of this unique trick. The carboxy terminus of NSP1 binds to the 40S ribosomal RNA, obstructing messenger RNA entry and translation. Simultaneously, NSP1 recognizes the stem-loop structure at the 5′ terminus of viral genomic and messenger RNAs, allowing viral mRNA ribosomal entry and translation.
Three other viral proteins collaborate to prevent cellular messenger RNAs from reaching the cytoplasm. The viral protein NSP16 inhibits messenger RNA splicing by binding to both ends of the RNA in the spliceosome. Unspliced cellular messenger RNAs remain in the nucleus and decay rapidly. Another viral protein, NSP9, prevents the export of cellular messenger RNAs from the nucleus.
Ending COVID-19
Two anti-SARS-CoV-2 drugs recently made headlines due to their promise in treating COVID-19. Both are antiviral oral pills. The first, called molnupiravir, developed by the pharmaceutical company Merck & Co., was authorized for use in Britain on November 4, 2021, after a global clinical trial found that it reduced the rate of hospitalization and death in COVID-19 patients by nearly 59%. However, as I’ve pointed out earlier (in “Supercharging New Viral Variants: The Dangers of Molnupiravir,” an article that appeared in Forbes), it appears, based on studies of other coronaviruses, that molnupiravir could have mutagenic effects on SARS-CoV-2 that might lead to the emergence of new and dangerous viral variants. This potential for supercharging viral variation currently remains overlooked by the regulatory agencies tasked with approving molnupiravir for widespread use.
The second pill was developed by Pfizer under the codename PF-07321332. On November 5, 2021, Pfizer announced that its pill reduced the risk of hospitalization and death in COVID-19 patients by nearly 90%. Although more data is needed, at this early stage, it doesn’t appear that the concerning potential for mutagenesis seen in the Merck pill is present in the Pfizer pill.
Looking forward, I am aware of dozens of antiviral drugs that are in early stages of either preclinical or early clinical trials. I am certain that over the next two years, we will have a broad array of pharmacologically active drugs safe enough to be used in combination. My vision is they will be safe enough to be preventives for those exposed and treatments for those infected.
Other anti-SARS-CoV-2 drugs approved or in advanced clinical trials include monoclonal antibodies, and drugs that target either NSP5 protease or the NSP12 polymerase (Figure 3, Table 1).
 Monoclonal antibodies targeting the spike protein provide proof of principle that antiviral drugs can prevent and treat vaccine breakthrough infections and infections of the unvaccinated. Although effective, they are expensive to manufacture, difficult to administer, and subject to escape mutants. The antipolymerase and antiprotease drugs are inexpensive to make and orally available.
Monoclonal antibodies targeting the spike protein provide proof of principle that antiviral drugs can prevent and treat vaccine breakthrough infections and infections of the unvaccinated. Although effective, they are expensive to manufacture, difficult to administer, and subject to escape mutants. The antipolymerase and antiprotease drugs are inexpensive to make and orally available.
Our experience with HIV teaches us that there are limits to the use of even these drugs. The first HIV drugs also inhibited the HIV polymerase and protease. The dose of these drugs is limited by toxicity; at high concentrations, they affect our own enzymes that carry out essential functions. Decades of experience taught us that a new generation of drugs was needed.
Today, the most successful anti-HIV drugs target viral proteins that have no human counterparts—the viral integrase and capsid. These drugs are many orders of magnitude less toxic. The hope is that one treatment every six months with such a drug combination is all that will be needed to prevent and treat HIV infections.
My own hope is that researchers, biotechnology companies, and pharmaceutical companies vigorously exploit the opportunities for drug development afforded by the wealth of viral targets discussed here, the very proteins SARS-CoV-2 requires to circumvent innate immunity. I foresee the day when a combination of vaccines and antivirals will ensure that no one becomes critically ill and dies of COVID-19.
William A. Haseltine, PhD, is the founder, chairman, and president of ACCESS Health International, a foundation dedicated to improving access to high-quality health services worldwide. He is known for his groundbreaking work on HIV/AIDS and the human genome, and he has founded more than a dozen biotechnology companies, including Human Genome Sciences.
Part 1 can be viewed in the January 2022 issue of GEN.

