
Sponsored content brought to you by
![]()
Viruses have evolved to invade host cells and hijack their cellular machinery to direct the replication and transcription of their own genomes. While these properties make viruses dangerous pathogens, they also make them ideal couriers for getting new DNA into a host cell and ensuring that it’s transcribed. Here, we describe three aspects of viral biology that make them efficient delivery vehicles for gene therapies, and some of the ways that OXGENE has taken advantage of these properties to improve clinical performance.
1. Viral entry into target cells
Adeno-associated virus (AAV) vectors are usually the vectors of choice for in vivo gene therapies. The cells they can infect, and how efficiently they do so, is defined by the specific cell surface receptors the viral capsid interacts with. So far nine natural AAV serotypes have been identified that infect human cells. Each
serotype has different tropisms that informs the choice of recombinant AAV (rAAV) most appropriate for each gene therapy.1
Despite the divergent tropisms offered by different AAV serotypes, efficient delivery of gene therapy vectors remains a challenge, either due to natural immunity to some AAV serotypes, limited tissue dispersion, or difficulty ensuring efficient delivery to a specific cell population. Capsid engineering offers an opportunity to improve transduction of the target cell type, improve specificity of transduction, and/or to evade a host immune response. There are generally two ways to approach this: rational design, which relies on understanding the mechanism of transduction and identifying the areas of the viral genome
responsible for encoding the capsid’s interaction with the cell surface receptor, and directed evolution, which is a more suitable approach when the cell surface receptors encoding the desired tropism are unknown.2
At OXGENE, we combine these approaches using proprietary bioinformatics to inform a rational approach to designing AAV genome plasmids containing a silenced capsid library alongside an eGFP reporter. We then use TESSA™ technology in a two-step process to build an AAV library where the AAV particles are encapsulated by the same cap protein they encode, which can be screened to identify novel functional capsids with the desired infectivity and specificity.
2. Viral genome expression
Once they’ve entered the cell, viruses are adept at wresting control of the cell’s DNA replication machinery to force the transcription of their own genomes. When the genes that would enable viral replication are replaced inside a virus particle by a therapeutic transgene, it is this DNA that the cell is forced to transcribe.
Almost all gene expression is driven by a promoter, a core region of DNA approximately 100 bp upstream of the regulated gene plus an upstream activating sequence or enhancer. Both core promoter regions and enhancers contain clusters of transcription factor binding sites, and transcription factor binding in these areas determines the level of gene expression.
Generally, either promoters endogenous to the target cell/tissue type or viral promoters are used to drive transgene expression. Tissue-specific endogenous promoters have the advantage of limiting gene expression to the target cells; however, typically low promoter activity or large promoter size may lead to suboptimal expression levels. Viral promoters, on the other hand, are constitutively active, resulting in high levels of therapeutic transgene expression in all transduced cells. This carries a risk of toxicity due to transgene overexpression or stimulation of an immune response due to transgene expression within an antigen-presenting cell.3
Engineering the promoter driving transgene expression therefore represents another opportunity to improve the clinical performance of the viral vector. By using proprietary bioinformatics to aid the rational design of combinatorial promoter/enhancer libraries, packaging these into lentiviral constructs containing a reporter gene and a unique index and barcode, and then infecting target cells at a very low multiplicity of
infection (MOI), OXGENE can screen for optimal promoter combinations that are designed to custom specifications. We can finely tune novel promoters to the required level for each gene of interest (GOI) or introduce novel regulatory properties to allow responsiveness to multiple biological signals.
3. Viral replication
Gene therapy manufacture relies on being able to produce huge quantities of viral vectors, using cells as mini virus-production factories. However, one of the properties of AAV that make it ideal as a vector for gene therapies—its inability to replicate without the help of a second virus—adds challenges for its manufacture. In nature, this help is provided by adenovirus, but this isn’t a system that’s easily co-opted for clinical production, because the adenoviruses must be purified out from the AAV before they’re suitable for clinical use, a complex and expensive process. To date, the adenoviral help necessary for producing AAV destined for clinical use is most often provided by a plasmid encoding an adenoviral genome that’s been pared back to only those genes essential for AAV replication. However, this is far from ideal, and the complexities and costs associated with plasmid-based AAV production at scale have been widely reported.
Adenoviruses, on the other hand, are extremely efficient at replicating themselves once they’ve gained control of the host cell systems. Indeed, following adenovirus infection, almost 90% of the host cells’ mRNA is derived from the adenoviral genome. OXGENE’s TESSA™ technology offers an opportunity to harness the power of adenoviral protein production and use it to improve the manufacture of AAV for gene therapies.
TESSA™ technology works by manipulating the AAV genome in such a way that it can still provide help for AAV production but can no longer transcribe adenoviral structural proteins. This takes advantage of the fact that the adenoviral genome is divided into two temporal phases with distinct purposes. All the genes responsible for helping AAV to replicate are transcribed in the early phase, while adenoviral structural proteins are all transcribed from late-phase genes, under the control of the major late promoter (MLP). Inserting a Tet repressor binding site into the MLP, and the Tet repressor gene itself into the late region of the genome, essentially cripples adenoviral production; the more the adenovirus tries to transcribe its structural proteins, the more Tet repressor it produces to inhibit MLP activity.
The next step is the insertion of either the AAV rep and cap, or the ITR flanked therapeutic transgene, into the early phase of the genome. Now, the adenoviral genome can no longer produce adenovirus, but infecting cells with two of these edited adenoviral vectors is sufficient to produce high quantities of fully functional, highly infectious AAV (Figure 1). So far, for every serotype tested, TESSA™ technology produces higher yields of AAV than triple transfection, and in most cases, this AAV is also more infectious, raising the possibility of a lower effective dose of the final gene therapy product, manufactured at a lower cost and with reduced process complexity.
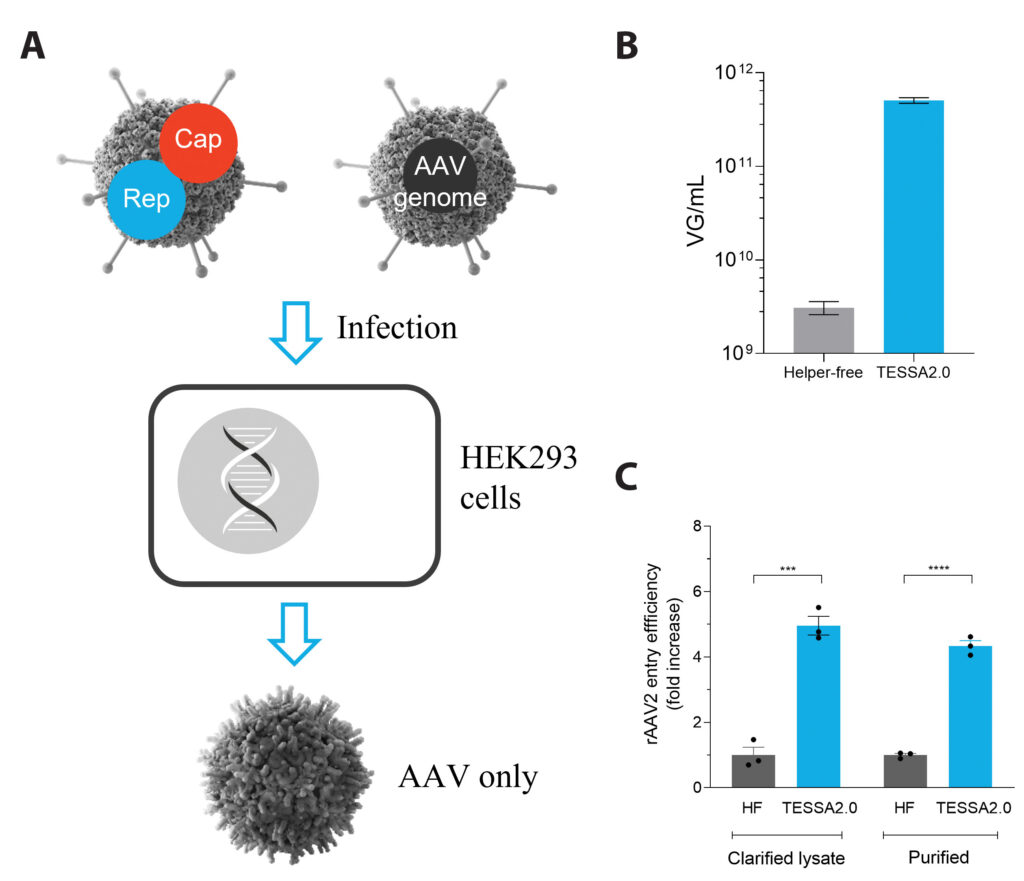
(C) rAAV2 produced using TESSA technology displays significantly better cell entry than rAAV2 made from triple transfection.
There are well over 1,000 cell and gene therapies in clinical trials ongoing worldwide, and even more in preclinical development. Recent advances in viral vector engineering, such as those described here, aim to accelerate the clinical success of these groundbreaking new therapeutics, while ensuring accessibility to patients through improved manufacturing processes with reduced costs.
For more information about OXGENE’s solutions, please visit www.oxgene.com
References
- Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016 Dec; 21: 75–80. doi: 10.1016/j.coviro.2016.08.003.
- Li C, Samulski, RJ. Engineering adeno-associated virus vectors for gene therapy.
Nat. Rev. Genet. 2020; 21: 255–272. Doi:10.1038/s41576-019-0205-4 - Blazek J, Alper HS. Promoter engineering: Recent advances in controlling transcription at the most fundamental level. Biotechnol J. 2013 Jan; 8(1): 46–58. doi: 10.1002/biot.201200120.