
A fundamental challenge in biology is precisely linking gene regulatory networks to the gene expression profiles that define unique cell types and states. In an effort to deeply characterize cell identity, researchers have historically looked to two separate assays that measure the transcriptome and epigenome in single cells. This requires two independent single cell and single nuclei partitions derived from the same tissue sample or cell line, followed by inferred linkage between separate gene expression and chromatin accessibility datasets.
Researchers can now capture both transcriptomic and epigenomic modalities simultaneously in the same single cells with Chromium Single Cell Multiome ATAC + Gene Expression. This technique removes the computational errors associated with linking separate datasets and maximizes possible readouts from precious samples. Moreover, with a unified view of a cell’s open chromatin landscape and gene expression profile, researchers can resolve how cell types and states are established, discover new gene regulatory interactions, and interpret epigenetic profiles with key expression markers.
What does it take to simultaneously profile the transcriptome and epigenome? Chromium Single Cell Multiome ATAC + Gene Expression comes with new experimental considerations for researchers versed in other techniques like single cell RNA-sequencing (scRNA-seq). Single nuclei are profiled instead of single cells. Data analysis includes exploration and interpretation of regions of open chromatin alongside gene expression readouts. The following sections explore the workflow, review data highlights, and offer tips for successful single nuclei sample preparation.
Overview of workflow and data output
The workflow begins with a suspension of nuclei containing DNA and nuclear mRNA. Transposition is performed in bulk upon application of the enzyme transposase, which enters the nuclei and preferentially fragments the DNA in open regions of chromatin. Transposed nuclei are loaded onto a microfluidic chip, which is run in the Chromium Controller. In this process, nuclei are partitioned individually into droplets with a single Gel Bead that contains a unique 10x Barcode. The droplets, or Gel Beads-in-emulsion (GEMs), are then incubated to attach the unique barcodes to mRNA and transposed DNA fragments from the same nuclei.
Following this incubation, GEMs are broken and pooled fractions are recovered and purified. The product is taken through a pre-amplification PCR step to fill gaps and ensure maximum recovery of barcoded ATAC and cDNA fragments. Subsequently, the pre-amplified product is used as input for both ATAC library construction and cDNA amplification for gene expression library construction (Figure 1). Resulting ATAC and gene expression libraries enable linkage of gene expression and open chromatin profiles back to the same cell with certainty.

Following sequencing, raw data is loaded into the Cell Ranger ARC analysis pipeline and visualized using Loupe Browser. Both software tools are available for download on the 10x Genomics Support website. Together, they provide a view of the linkage of single cell ATAC and gene expression data across a population of cells and in specific cellular clusters.
Linkage of transcriptomic and epigenomic modalities indicates when the expression of a certain gene correlates with specific open chromatin regions around it (Figure 2). Detection of feature linkage reveals how open chromatin might affect gene expression in a specific cell type or cell state, relative to other cell types in a diverse population. A positive linkage between gene expression and an open region of chromatin, or a chromatin peak, suggests that this peak may play an activating role in the expression of the gene.
Conversely, the presence of a certain peak in a subset of cellular clusters may be strongly correlated with the absence of expression. This indicates the peak may play a role in repressing gene expression.
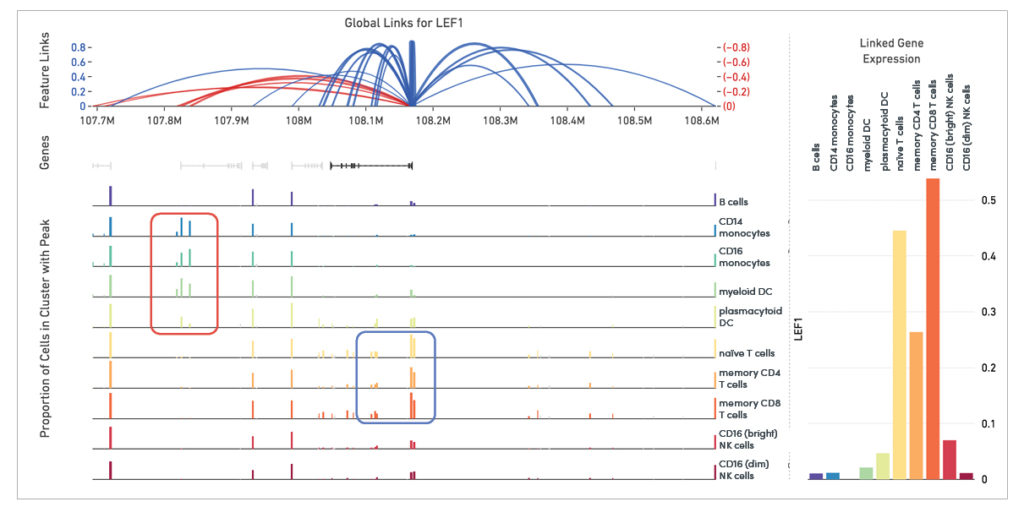
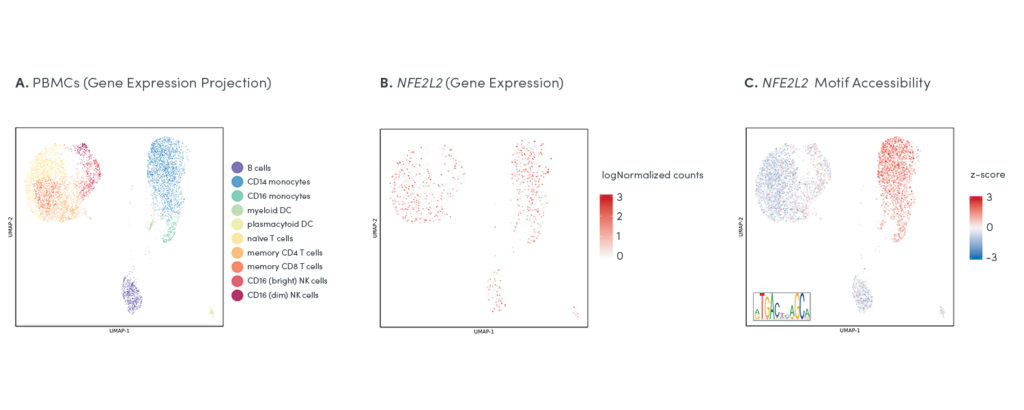
The ability to map gene expression and chromatin accessibility within the same cells gives researchers an opportunity to use two data types to inform deeper cellular annotation, and make new discoveries about the functional role of key regulatory elements at work in specific cell types and states. Tracking the differences between gene expression and chromatin state for regulatory elements in the same cells can shed new light on the function of certain transcription factors in specific populations (Figure 3).
Best practices for single nuclei sample preparation
Successful nuclei isolation requires attention to detail. Depending on a number of factors–including sample type, whether you are working with whole tissue or a cell suspension, whether you are starting with fresh or frozen materials, the manner in which the sample was frozen, and cell count—there will be specific decision points in the nuclei isolation protocols that change how the sample is handled.
10x Genomics has developed specialized protocols optimized for multiomic profiling and offering guidance for each of these factors. Explore a few key decision points and associated best practices from our nuclei isolation protocols in the sections below.
Fresh or frozen cells
Fresh cells can be washed, counted, and then moved forward to nuclei isolation steps. Frozen cells will require additional thawing steps. Thawing steps will also differ slightly for primary/fragile cells, including PBMCs or bone marrow mononuclear cells (BMMCs), due to potentially high levels of background DNA. One option to alleviate this is to sort cells before performing nuclei isolation, which will also allow researchers to enrich for their cell types of interest.
Nuclei isolation
If cell count is low, or less than 100,000 cells, an alternate protocol is provided for nuclei isolation. No matter the cell count, there are a few important steps to keep in mind. Cell lysis time is optimized based on sample type. Lysis efficacy is then assessed using a high power microscope. Optimal nuclei isolation is reached when most cells stain as dead, meaning the cell membrane has broken, but the nuclear membrane looks intact.
After washing, isolated nuclei are resuspended in chilled Diluted Nuclei Buffer at a stock concentration dictated by targeted nuclei recovery counts. Diluted Nuclei Buffer is critical for optimal assay performance, including the transposition and barcoding steps of the workflow.
From start to finish, find resources to navigate each new consideration for your Chromium Single Cell Multiome ATAC + Gene Expression experiment, including single nuclei isolation protocols, at support.10xgenomics.com/single-cell-multiome-atac-gex.
Kamila Belhocine, PhD, is a staff scientist, Laura DeMare, PhD, is a product manager, and Olivia Habern is a marketing content writer at 10x Genomics.