
March 1, 2010 (Vol. 30, No. 5)
Automating and Parallelizing the Process Can Help to Avoid an Analysis Bottleneck
Pharmaceutical and biomedical research is evolving to take advantage of the development of bioinformatic research programs, incorporating data from new high-resolution assays and technologies such as microarrays and fluorescent in situ hybridization.
Information supplied by these methods concerning the physiological functions of genes can provide a molecular understanding of the mechanism of diseases, which can lead to effective therapies. It can also assist in basic scientific research, such as improving our understanding of genetic networks or embryological development.
The value added by these new assay technologies resides in the detailed insight provided by the high-resolution data they output. This blessing, however, is also a curse: the huge volume of data must be processed, assessed for quality, and have its relevant features extracted before any visualization or statistical analysis is possible.
The time when it was possible for a lab scientist to manually perform this processing has now passed; researchers would prefer to put their education and skills to use by concentrating on the science, rather than wasting their days repetitively annotating images or manipulating tables in a spreadsheet program. Manual processing is also error-prone and often subjective.
For these new technologies, then, automated processing is essential. Effective development of data-processing algorithms demands tools that enable rapid prototyping and implementation of these algorithms; ideally this will be the same environment that is subsequently used for visualization and statistical analysis of the processed data.
Automated processing is a first step only, however. The deployment of assay technologies in a high-throughput environment can mean that even an automated data-processing step will be the bottleneck. In this case, researchers can take advantage of the increasing availability of computer clusters to parallelize their data processing, releasing the bottleneck so that research can take place at the speed of science, not of analysis.
We explore these issues further in a case study—the automated parallel-processing and subsequent visualization and analysis of data from the FlyEx database.
The FlyEx database is a web repository of segmentation gene-expression images. It contains images of fruit fly (Drosophila melanogaster) embryos at various stages of development (cleavage cycles 10–14A) and quantitative data extracted from the images. Such work is invaluable in understanding the developmental processes in the embryo. Spatial and temporal patterns of gene expression are fundamental to these processes.
The images are created using immunofluorescent histochemistry. Embryos are stained with fluorescently tagged antibodies that bind to the product of an individual gene, staining only those parts of the embryo in which the gene is expressed. In this case, the genes studied are those involved in the segmentation of the embryo, such as the even_skipped, caudal, and bicoid genes. The embryo is then imaged using confocal microscopy.
The database currently contains more than two million data records and thousands of images of embryos. Clearly, the automated processing of these images is a necessary step in the construction of the database.
Mathworks has developed an algorithm for processing the images using MATLAB and Image Processing Toolbox.
MATLAB is an environment for technical computing, data analysis, and visualization. As an interactive tool, it enables researchers to prototype algorithms and analyses; its underlying programming language, optimized for handling large scientific datasets, allows these prototypes to be automated for high-throughput applications. Toolboxes provide application-specific add-on functionality, such as image and signal processing, multivariate statistics, and bioinformatics.
The image-processing algorithm involves a number of steps. The embryo image is first rotated into a standard configuration—centered, with the anterior-posterior axis horizontally oriented. The second step removes noise from the image and equalizes the brightness across the entire image using adaptive histogram equalization.
Finally, the boundaries of each cell in the embryo are found using a brightness thresholding step, and the pixels within each cell boundary are separated into their red, green, and blue channels. These red, green, and blue channels correspond to the level of expression of these three segmentation-related genes: even_skipped, caudal, and bicoid. Figure 1 shows a profile of the three expression levels along the anterior-posterior axis.
The algorithm is available from MATLAB Central, an online community that features a newsgroup and downloadable code from users at the File Exchange. The use of prebuilt methods, such as brightness thresholding, histogram equalization, and median filtering for noise reduction (supplied as building-block algorithms in Image Processing Toolbox), together with the ability of MATLAB to treat images simply as numerical arrays, allow the prototype to be compact, readable, and rapidly constructed.
The processed results can then be analyzed and visualized using the same MATLAB environment. This approach offers important advantages in reducing the number of tools necessary for the overall analysis—streamlining the process, reducing the training needs of scientists, and removing a source of error in the transfer of data to a separate statistical package.
The results could be analyzed according to the nature of the experimental context. For example, you could use:
- time-series methods to examine how the gene-expression profiles vary over time;
- ANOVA to examine the effects of different treatments or disease conditions; or
- multivariate statistical or machine-learning methods, such as Principal Component Analysis or Cluster Analysis, to examine complex relationships within the profiles and between samples.
Analysis results can also be visualized with other data sources and annotations, such as publicly available information on the genes themselves, public repositories of genetic data such as GenBank, and genetic pathway visualizations.
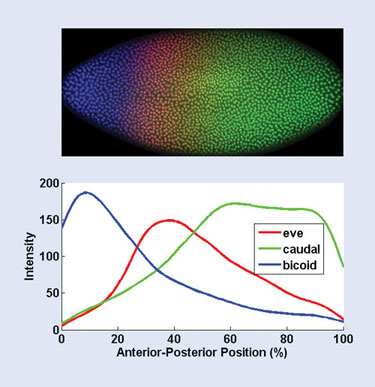
Figure 1. Results of the automated image-processing and statistical analysis of a fruit-fly embryo
Automation
The prototype algorithm described above automates the processing of a single image. By taking advantage of the underlying programming language of Matlab, it is a simple matter to apply this algorithm in a batch process to analyze thousands of images automatically, or in a continuous process to analyze images as they are generated by a high-throughput process.
Depending on particular IT infrastructure, generated image data might be accessed by MATLAB from files on disk, from a database, or even directly streamed in from the microscopes themselves. Processing and analysis results can be output to a report file, to a database, or perhaps to a web page.
Parallel Computing
Depending on the nature of the particular steps taken in the image-processing algorithm, an individual image could take from a few seconds to a minute to be processed on a typical desktop computer. Processing the few thousand images in the FlyEx database might then take from half a day to a few days, a time frame that perhaps is acceptable in the context of a research program in developmental biology.
In a pharmaceutical context, however, a typical high-throughput screening library contains one or two million compounds that are tested by an HTS robot at a rate of up to 100,000 compounds per day. A time frame of several months to process these generated images would be an unacceptable delay.
Parallel Computing Toolbox enables scientists to solve computationally intensive problems using MATLAB on multicore and multiprocessor computers, or scaled to a cluster, using Distributed Computing Server. By distributing the processing of high-throughput screening data across multiple computers, researchers can decrease analysis time by orders of magnitude.
Simple parallel-programming constructs, such as parallel for-loops, allow scientists to convert algorithms to run in parallel with minimal code changes and at a high level without programming for specific hardware and network architectures (Figure 2).
It is crucial that scientists are able to easily convert their algorithms to work in parallel while remaining at this high level, without needing to become experts in the traditionally complex techniques of programming for high-performance computing.

Figure 2. The parfor (a parallel for-loop) keyword from Parallel Computing Toolbox enables simple conversion of a batch-processing application to run in parallel, taking advantage of a compute cluster.
Future Trends and Requirements
It is already a clichéd concept that the new technologies currently expanding the boundaries of pharmaceutical and biomedical research are generating ever-increasing amounts of data, and that automated analysis algorithms and efficient use of parallelization using computer clusters are vital if we want to avoid an analysis bottleneck and allow science to proceed at its own rapid pace.
Tools are, therefore, needed to enable the prototyping, automation, and easy parallelization of these algorithms. The use of technical computing environments such as those described in this article allow scientists to move rapidly from interactively visualizing data, to prototyping a processing algorithm, to automation, to a high-throughput solution in a single environment.
Adoption of these techniques will allow science to avoid analysis bottlenecks and to continue to proceed at its own pace, even in a high-throughput context.
Sam Roberts ([email protected]) is principal application engineer at MathWorks. Web: www.mathworks.com.

