
Peptide therapeutics would seem to occupy a sweet spot between small molecules and biologics. Small molecules, which represent the majority of clinical drugs, preferentially bind hydrophobic pockets or cavities, but targeting them to protein interfaces that are large or shallow, or contain combinations of polar and apolar residues, is challenging.
Biologics, or larger molecules, use more extensive contact areas and can modulate protein–protein interactions extracellularly, but are difficult to deliver into cells.
The difficulties posed by small molecules and biologics could be avoided by peptide therapeutics, which present drug developers with distinct advantages. These include simpler production, lower costs, and superior safety and tolerability. Consequently, some drug developers see peptide therapeutics as an attractive means of reaching targets that are known to be therapeutically relevant but are, as yet, “undruggable.” Peptide therapeutics, however, need to manifest sweet spots of their own.
For example, peptide therapeutics need to overcome the limitations of naturally occurring peptides, which include low bioavailability, physical and chemical instability, low membrane permeability, short half-life, and fast elimination. A peptide’s limitations may be addressed via chemical modifications, but such modifications shouldn’t attenuate the peptide’s strengths, which include specificity, potency, safety, predictable metabolism, and ease of synthesis. This is a balancing act, and it is performed not only for modified versions of natural peptides, but also for peptides that possess novel structures, or are abstracted or excerpted from larger proteins.
Permeability First
“We are focused on targeting intracellular protein–protein interactions that are key drivers in oncology pathways,” says David J. Earp, Ph.D., president and CEO of Circle Pharma. Research at Circle Pharma is focusing on macrocycles, which are cyclic molecules of at least 12 atoms and are favorably positioned for targeting protein interfaces that are difficult to interrogate.
The polar surface areas of macrocycles, however, present challenges with respect to cell permeability and oral bioavailability. “We found a very good solution for the permeability issue,” asserts Dr. Earp, who observes that poor permeability is common problem in the development of macrocycle drugs.
The discovery platform at Circle Pharma combines automated synthetic chemistry with computational permeability design and medicinal chemistry–oriented structure-based drug design for peptide–peptoid hybrid macrocycles. “What makes our platform attractive is that permeability is the very first thing that we build into the design process, and therefore it is not something that we have to solve later on during development,” explains Dr. Earp, “and the peptoids in our macrocycles enable wide chemical diversity, well beyond peptide macrocycles.”
In a recent collaboration with Pfizer, Circle Pharma developed a potent and orally bioavailable macrocyclic peptide targeting CXCR7, a chemokine receptor clinically attractive for applications that include oncology and autoimmune, metabolic, and infectious diseases. This work originated from previous research that found a hexamer peptide macrocycle with modest CXCR7 binding affinity and no measurable cell permeability (and therefore no oral bioavailability).
“Using our computational design platform, we created cell-permeable derivatives of that initial macrocycle,” notes Dr. Earp. “By incorporating and exploiting peptoids, we were able to achieve 500-fold improvement in target affinity.” In rodent studies administered by Pfizer, one of these compounds, demonstrated 18% oral bioavailability.
“In addition to creating derivatives of the initial macrocycle, we also successfully scaffold-hopped, screening our large virtual libraries to identify new scaffolds with good permeability characteristics and target fit,” continues Dr. Earp. “We then advanced affinity using structure-based medicinal chemistry.”
The chemistry-extensive automated platform allows investigators at Circle Pharma to produce macrocycles rapidly and at a low cost, allowing several design hypotheses to be tested in parallel. These investigators are focusing on so-called “undruggable” oncology targets for which high-quality structural data is available to facilitate drug design.
“We are working on compelling targets that are well understood from the biology perspective but have not been clinically validated,” says Dr. Earp. “As our programs advance, we are starting to look at metabolic liability in our macrocyles. It appears to be similar to small molecule drug metabolism, and we can modulate it with appropriate side-chain substitutions.”
Scientists at Circle Pharma are working to develop a deeper understanding of metabolic liability, one that they can use to refine their computational algorithms and improve their design methods. “At the same time, we are actively reviewing our target selection while taking into account newly available structural information and scientific advances,” declares Dr. Earp. “As we expand our platform throughput with further synthesis automation, we will have the ability to work on multiple targets in parallel while we add or switch targets as appropriate.”
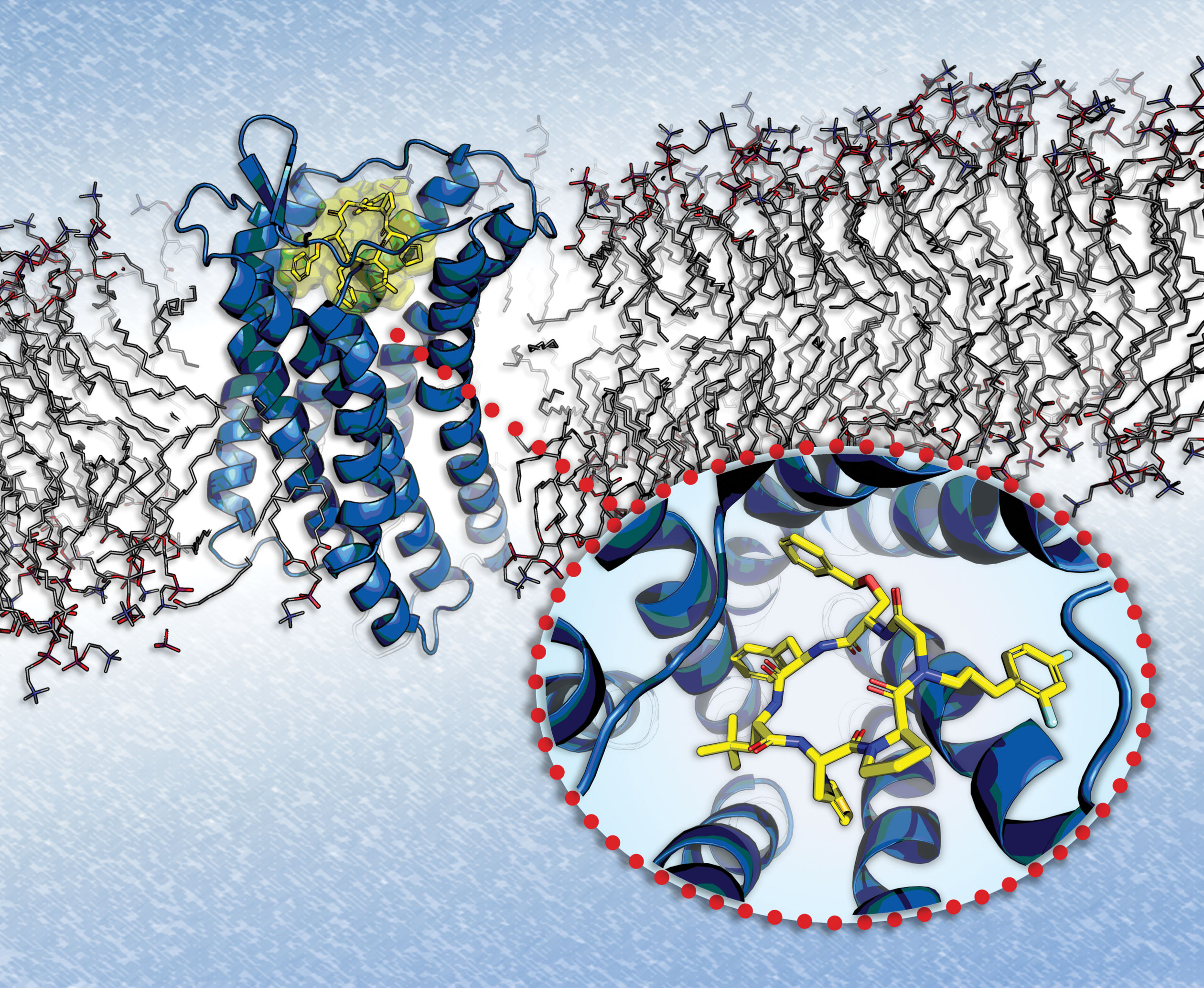
CXCR7, a chemokine receptor implicated in several diseases, is being targeted by peptidic macrocycles produced by Circle Pharma. These molecules incorporate a residue that permits extensive exploration of peptide side chains. Models and assays lead to peptidic macrocycles, such as the one shown here (yellow), that may perform better than small molecules in terms of potency and selectivity.
Complement Immunotherapy
“With APL-2 we prevent the very first step of complement deposition on cell surfaces,” says Cedric Francois, M.D., Ph.D., co-founder, CEO, and president of Apellis Pharmaceuticals. A key effort at Apellis involves the development of complement immunotherapies, a strategy that shows promise for treating autoimmune and inflammatory conditions in which the complement cascade orchestrates the pathogenesis.
When complement components are targeted, complications may ensue because complement components are involved in many cellular processes. “This is why there were a lot of fears that inhibiting C3 may lead to unintended consequences,” explains Dr. Francois.
APL-2, the lead molecule developed by Apellis, is made of two synthetic cyclic peptides conjugated to a polyethylene glycol polymer. APL-2 binds specifically to a pocket in C3 and C3b and inhibits all three pathways of complement activation.
Using APL-2, scientists at Apellis developed two clinical programs: a systemic program and a local program. These programs, which are being run in parallel at Apellis, are highly complementary, maintains Dr. Francois, who adds that they are both generating “very promising data.”
In the systemic program, APL-2 is injected subcutaneously to inhibit C3 systemically, and the lead indication is for paroxysmal nocturnal hematuria. “The systemic indication is a great proof of concept, but the challenge is to make sure it is safe,” notes Dr. Francois.
In the local program, which was developed for ophthalmological applications, a very small amount of the therapeutic is injected into the eye. “Safety is far less of a consideration for the ophthalmological application, but establishing efficacy is a much bigger hurdle,” indicates Dr. Francois.
The ophthalmological application of APL-2 is for geographic atrophy, one of the two advanced forms of macular degeneration, the other one being wet age-related macular degeneration. Geographic atrophy is characterized by the progressive destruction of the retina starting from the periphery. The areas of atrophy make the retina appear like a map upon examination, explaining the term given to this condition.
If the fovea is spared, which is typical of the initial phase of the disease, visual acuity may remain stable over time even though the visual function progressively becomes impaired. However, at advanced stages, once the fovea is affected, visual acuity is lost dramatically over a very short time.
“The best analogy is with a forest fire,” suggests Dr. Francois. The lesion is initially localized to a small area of the retina. “But eventually,” Dr. Francois points out, “the entire retina disappears.” Any therapy that could slow the rate of retinal destruction would be highly beneficial.
“Because of that progression, for Phase III trials of geographic atrophy, the FDA has allowed investigators to use not visual acuity but, instead, the growth of atrophy as the primary endpoint,” says Dr. Francois. The registration endpoint involves measuring the area of retinal atrophy, which is indicative of the rate of photoreceptor loss
“This is something that we have very convincingly shown in our Phase II trial,” asserts Dr. Francois. In this trial, APL-2 was administered as an intravitreal injection in the study eye monthly or every other month for 12 months, followed by six months of monitoring after the end of treatment.
“At 12 months, APL-2, administered monthly via intravitreal injection, showed a 29% reduction in the rate of GA lesion growth compared to sham,” the company indicated in a press release. “With every other month administration, a 20% reduction was observed. Additionally, in a post hoc analysis, a greater effect was observed during the second six months of the study: a reduction in growth rate of 47% with monthly administration, and a reduction of 33% with every other month administration.”
According to Dr. Francois, this trial demonstrates that complement immunotherapy can slow photoreceptor loss in patients with geographic atrophy, a disorder that affects 1 million people in the United States. No treatments for this disorder are currently available on the market, and all other leads in clinical development have failed. “We look forward to our Phase III clinical trials to test whether we can repeat the data from the Phase II trials,” states Dr. Francois.
Preventing Amyloid Deposition
“Many of the approaches to reduce amyloid-β (Aβ) peptide have failed not because scientists were unable to reduce the levels of the peptide, but because their approach—the targeting of the enzymes that cleave Aβ from its precursor—can lead to undersirable side effects,” says Nazneen Dewji, Ph.D., president and CEO of Cenna Biosciences, and associate adjunct professor of medicine at University of California, San Diego. Aβ, the major neurotoxic agent in Alzheimer’s disease, and the main component of the amyloid plaques that are found in the brains of patients with Alzheimer’s disease, is formed by the sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretases. Inhibiting the catalytic activity of either of the two enzymes inhibits the release of the Aβ peptide.
“However, each of these enzymes has 50–60 different substrates besides APP, and inhibiting these reactions, some of which are incredibly important to the cell, gives rise to many off-target effects,” says Dr. Dewji. For example, γ-secretase also catalyzes the release of the intracellular domain of Notch, which subsequently translocates into the nucleus to bind transcription factors and modulate gene expression. While most of the γ-secretase inhibitors designed for Alzheimer’s disease have failed, some of the β-secretase inhibitors, which have fewer adverse effects, are still being pursued.
“Our approach uses peptides that are derived from presenilin-1 (PS-1), which is one of the components of the γ-secretase complex,” says Dr. Dewji. The technology that Dr. Dewji and colleagues use is based on their observation that binding of a fragment of the amino terminal domain of PS-1 to APP can inhibit the production of Aβ. This binding to APP occurs at a location that is away from the β- and γ-secretase cleavage sites.
“This approach is incredibly specific,” asserts Dr. Dewji, who adds that the catalytic activities of the enzymes are “not targeted and remain intact.” Using the lead candidate peptide, investigators at Cenna Biosciences reduced Aβ levels in vitro and in transgenic mice by 50–70%. “We are moving forward with the development of this peptide,” Dr. Dewji declares. “Early next year we hope to start Phase I trials.”
The peptide can be delivered subcutaneously and it crosses the blood–brain barrier. “While an oral drug is always easier to administer,” notes Dr. Dewji, “subcutaneous administration is still a relatively easy route of administration compared to intravenous delivery.”
Modulating Metabolism
“Our story began when we observed what was happening in the clinic with patients undergoing bariatric surgery, and then we went to the preclinical stage to select our molecule, and from there we went back into the clinic,” recalls Cristina M. Rondinone, Ph.D., vice president of research and development and head of cardiovascular, renal, and metabolic diseases at MedImmune.
A key research effort at MedImmune is focusing on peptide therapeutics to treat diabetes and obesity. While existing therapeutic options may be helpful, none of them are curative. A critical observation was that the glucose metabolism improvements that occur after bariatric surgery precede the weight loss seen in patients.
“This indicates not only that something happened very fast, but also that the mechanisms for improving glycemic control are different from the ones to reduce body weight,” maintains Dr. Rondinone. As a consequence of bariatric surgery, the duodenum is bypassed, and there is a change in the secretion of peptides, hormones, and other biologically active molecules by the intestine. “We were interested in investigating which of those molecules were changing, and which of the peptide combinations would cure diabetes,” says Dr. Rondinone.
Scientists at MedImmune interrogated combinations of peptides and hormones and identified a peptide dual agonist of glucagon-like peptide-1 (GLP-1) and glucagon receptors. The peptide dual agonist mimics the effects of oxyntomodulin, a glucose- and appetite-regulating gut hormone. In patients with type 2 diabetes, the secretion of oxyntomodulin is significantly impaired, and after gastric bypass surgery, the oxyntomodulin level is increased by more than 10-fold.
The scientists reasoned that a modified version of oxyntomodulin might have therapeutic potential against obesity and diabetes. “We synthesized more than 500 peptides and changed the ratio between the GLP-1 and the glucagon receptor agonism,” details Dr. Rondinone. This work, he continues, “helped us find the ideal peptide,” that is, one capable of combining GLP-1’s benefits (on glucose homeostasis), as well glucagon’s benefits (on lipids and body weight). This is how our molecule was born,” says Dr. Rondinone.
A key component of this work involved peptide chemistry to develop and modify the peptides to make them more potent and extend their half-lives. For example, coupling to palmitic acid facilitated the reversible binding of the lead peptide to albumin and extended its plasma half-life.
Using systems biology data modeling and pharmacokinetic and pharmacodynamics screenings in animal models, Dr. Rondinone and colleagues selected MEDI0382 for clinical studies. MEDI0382 showed robust anti-obesity and metabolic effects in mice and nonhuman primates, and a randomized, double-blind, placebo-controlled Phase II trial on overweight and obese adults with type 2 diabetes mellitus is underway. “We have other peptides in our pipeline that might be beneficial for other indications apart from diabetes,” adds Dr. Rondinone.




