
Mindy I. Davis Ph.D. National Institute of Health
Researchers develop a direct-binding assay that enables the detection of allosteric inhibitors, which can suppress a cellular enzyme that contributes to leukemia.
ASSAY & Drug Development Technologies offers a unique combination of original research and reports on the techniques and tools being used in cutting-edge drug development. The journal includes a “Literature Search and Review” column that identifies published papers of note and discusses their importance. GEN presents one article that was analyzed in the “Literature Search and Review” column, a paper published in the Journal of the American Chemical Society titled “Direct binding assay for the detection of Type IV allosteric inhibitors of Abl.” Authors of the paper are Schneider R, Becker C, Simard JR, Getlik M, Bohlke N, Janning P, and Rauh D.
Abstract from Journal of the American Chemical Society
Abelson (Abl) tyrosine kinase is an important cellular enzyme that is rendered constitutively active in the breakpoint cluster region (BCR)-Abl fusion protein, contributing to several forms of leukemia. Although inhibiting BCR-Abl activity with imatinib shows great clinical success, many patients acquire secondary mutations that result in resistance to imatinib. Second-generation inhibitors such as dasatinib and nilotinib can overcome the majority of these mutations but fail to treat patients with an especially prevalent T315I mutation at the gatekeeper position of the kinase domain. However, a combination of nilotinib with an allosteric type IV inhibitor was recently shown to overcome this clinically relevant point mutation.
In this study, we present the development of a direct binding assay that enables the straightforward detection of allosteric inhibitors which bind within the myristate pocket of Abl. The assay is amenable to high-throughput screening and exclusively detects the binding of ligands to this unique allosteric site.
Commentary
Imatinib, a type II kinase inhibitor, has been very successful at treating chronic myelogenous leukemia (CML) caused by a fusion of Abelson (Abl) kinase with the breakpoint cluster region (BCR) that renders Abl constitutively active. However, over time mutations can arise that diminish or eliminate the effect of imatinib on the cancer cells.
One such mutation involves the replacement of the small gatekeeper residue with a bulky residue (T315I), thereby sterically blocking imatinib and the majority of second-generation inhibitors from the active site. This active-site mutation has been particularly hard to overcome and indicates the importance of continued research to find the next generation of CML drugs. Wild-type Abl is regulated by its Src homology domains (SH2 and SH3) and an N-terminal myristoylated region. This regulation is lost upon the fusion with BCR, which leads to a change in the conformation of an α-helix thereby blocking the SH2 domain from inhibiting Abl.
It was discovered previously that inhibitors can bind to the distal myristoyl site causing a 90° bend in the nearby α-helix, which once again allows the SH2 domain to inhibit the kinase domain of Abl. The initial discovery of a compound that acted on this distal site was by chance, and the goal here was to develop an assay that would specifically select for inhibitors of this distal site.
The authors used an assay format called FLiK (fluorescent labels in kinases), which had been previously applied to the activation and “glycine-rich” loops of kinases, to probe the distal myristoyl site. FLiK involves covalent modification of the protein with an environmentally sensitive fluorophore (acrylodan) at a site that will undergo a large conformational change upon compound binding (see Figure 1).
Prior to the covalent modification, mutagenesis of the protein was required. A cysteine residue was added to which the fluorophore was later linked (V338C), and other surface accessible cysteine residues were mutated (C305V and C330S) so that they would not be covalently modified. The assay involved monitoring the decrease in fluorescence upon compound addition due to the change in the environment of the fluorophore (see Figure 2).
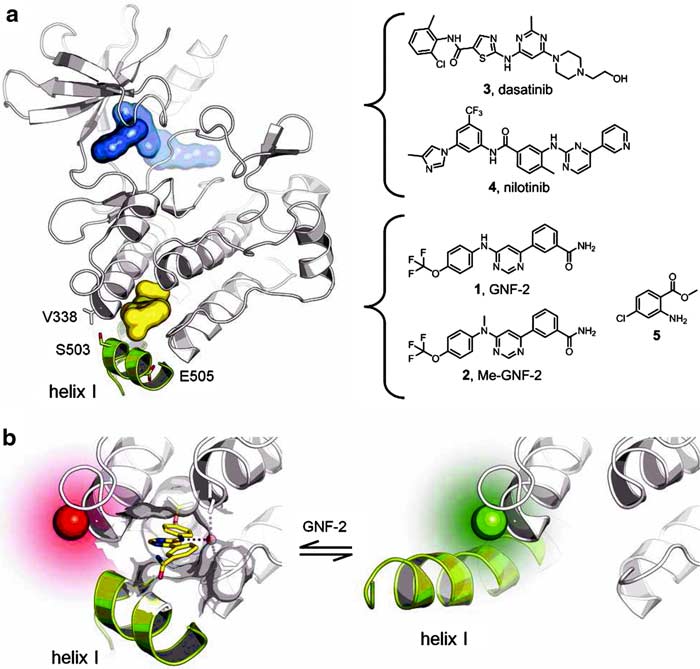
Figure 1. Schematic representation of Abl. (a) The kinase domain of Abl is shown with inhibitors addressing the active site (blue) and the myristate pocket (yellow). Labeling positions analyzed in this study are indicated (sticks). (b) Helix I is bent in the inactive conformation of Abl (left, PDB code 3k5v), while being straight in the active one (right, PDB code 2z60). This conformational change is reported by the attached fluorophore (spheres).
Figure 2. FLiK assay. (a) Stepwise addition of GNF-2 to acrylodan-labeled Abl (C305V/C330S/V338C) results in a significant and dose-dependent change in the fluorescence. (b) The Kd can be determined from plotting the logarithmic concentration of GNF-2 against the ratio of the two acrylodan emission peaks. No significant change in the ratio is observed for Me-GNF-2 and other control inhibitors. Due to intrinsic compound fluorescence, titration of 3–5 did not exceed 10 µM. (c) The binding of GNF-2 is fast and reversible. (d) Addition of vehicle alone had no effect.
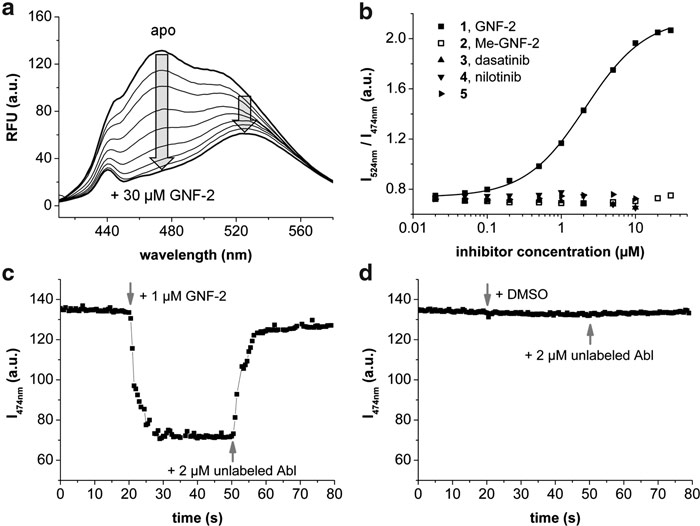
Figure 2
The 384-well assay had excellent statistics and was able to discriminate between inhibitors that bind to the active site in a Type I or Type II fashion and those that solely impact the myristoyl site; i.e., allosteric Type IV inhibitors. The allosteric Type IV pyrimidine scaffold inhibitors, such as GNF-2, show a lot of promise as CML drugs because they can inhibit the recalcitrant BCR-Abl(T315I) when coadministered with imatinib or nilotinib. It will be interesting to see what sort of new chemical starting points this assay design may uncover when screened against large libraries.
Mindy I. Davis, Ph.D., works at the NIH.





