
February 15, 2015 (Vol. 35, No. 4)
Software for Auto-Analysis of Internal Benchmarking Controls
As next-generation sequencing (NGS) makes genetic testing for a wide range of human diseases increasingly commonplace, facile methods for validating the efficacy of those tests are essential. Federal regulatory standards embodied in the Clinical Laboratory Improvement Amendments (CLIA), for instance, are designed to ensure that these tests reliably achieve certain performance specifications in terms of accuracy, precision, and analytical sensitivity and specificity.
To facilitate the test validation process, the National Institute of Standards and Technology (NIST) through the Genome in a Bottle Consortium (GIAB) developed a highly curated set of genome-wide reference materials for the HapMap/1000 Genomes CEU female, NA12878. These materials include BED and VCF files of high confidence sequence regions and variant calls, respectively. NA12878 genomic DNA and a cell line are available (Coriell Institute) providing laboratories with an internal control for their processing and analysis pipeline.
Comparing testing results to the GIAB call sets allows establishment of both the analytical performance for regulatory certification as well as the appropriate assembly thresholds to apply when considering potential variants in clinical samples.
Computational Challenge
For clinical sequencing laboratories to efficiently leverage these resources, assembly and analysis software must support the unique aspects of validation control processing. These include intersecting the GIAB high confidence regions and variant calls with the targeted genes of any given test, and matching the GIAB call set conventions wherever possible to determine the true accuracy of the results.
Additionally, data processing and statistical calculations should be rapid, automated, and reported in an easily interpreted form. Most clinical laboratories lack the bioinformatics expertise needed to build software capable of handling these challenges.
Software Workflow
DNASTAR has developed a dedicated workflow within its Lasergene Genomics Suite for clinical sequencing labs to exploit the NA12878 reference materials to validate their NGS-based genetic tests (Figure 1).
Within the SeqMan NGen® setup wizard, users specify: 1) NGS reads from their processed NA12878 sample; 2) the human genome reference version (NA12878 reference materials are in GRCh37 coordinates); 3) an intersected BED file between their targeted regions and GIAB’s NA12878 high confidence regions; and 4) GIAB’s NA12878 VCF file of high confidence variant calls. At runtime, the VCF is filtered down to those positions delineated by the intersected BED file.
The data is then assembled against the human genome reference sequence using SeqMan NGen running on a standard desktop computer. Fully gapped alignments are analyzed in-stream using a modified version of the MAQ variant caller to produce variant and reference call files for each position in the intersected BED. Key metrics, including the depth of coverage and probability scores, are recorded for each position. Assemblies can be visualized in SeqMan Pro allowing evidence for variants and regions of low coverage to be assessed.
For accuracy calculations, variant and reference call files are loaded with the filtered VCF file into ArrayStar®. Only positions within the intersected BED are considered. Positions are classified as: 1) true positives (TP), called variants also present in the VCF file; 2) false positives (FP), called variants not in the VCF file; 3) true negatives (TN), called reference bases not in the VCF file; and 4) false negatives (FN), called reference bases that are present in the VCF file. The counts of each class are then used to calculate various accuracy metrics, including the true positive rate (TPR, “sensitivity”), the true negative rate (TNR, “specificity”), and the false discovery rate (FDR).
A summary report is produced with the absolute number of positions in each class and the corresponding statistics stratified based on two thresholds: 1) minimum depth of coverage, which a position must match or exceed to be considered in the analysis; and 2) minimum p-value a position must have to be considered a variant. Three p-values (“Pnotref” of 90.0%, 99.0%, and 99.9% corresponding to phred-like scores of 10, 20, and 30, respectively) are employed for each of 12 different depths of coverage cutoffs (ranging from 1 to 100). The number and percentage of targeted bases meeting each depth cutoff are also presented.
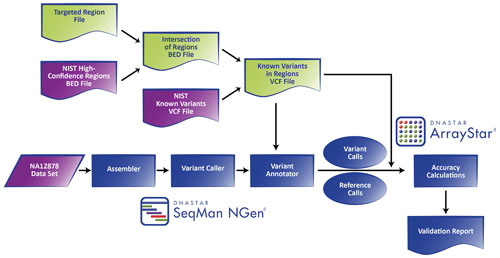
Figure 1. Data flow diagram
Case Study: Using NA12878 to Validate a Kidney Disease Gene Panel Test
ephropathology Associates (Nephropath™), the largest renal biopsy laboratory in the U.S., is in the process of developing a NGS-based test to aid in diagnosis of patients with idiopathic kidney disease. The test interrogates 301 genes involved in 13 different classes of renal disease. Genomic DNA from NA12878 and patient samples are processed in parallel using custom oligo-based capture followed by paired-end sequencing on an Illumina MiSeq instrument. After assembly and variant calling by SeqMan NGen, the NA12878 control sample is automatically loaded, processed, and validation accuracy statistics calculated in ArrayStar.
An excerpt of the report from a run of NA12878 through the Nephropath gene panel illustrates how the validation workflow can be used (Table). First, the calculated statistics and coverage information form the basis for intra- and inter-run performance characteristics required by regulatory programs such as CLIA. Second, the reported metrics can aid pathologists in balancing the trade-off between maximizing true positive detection and minimizing false positive calls to determine the appropriate cutoffs for patient sample variant calling.
As expected, increasing the depth requirement reduces both the number of true and false positives detected, while increasing the minimum p-value again reduces the number of true positives and increases the number of false negatives (e.g., a variant with a p-value of 99.1% moves from a true positive to a false negative when the threshold is increased to 99.9%). These changes are reflected in shifts in the class I and class II error measurements, TPR and FDR. The degree to which the targeted region has adequate depth of coverage is also important as regions with insufficient coverage must be reported and can point to problematic regions in the capture or sequencing that should be addressed.
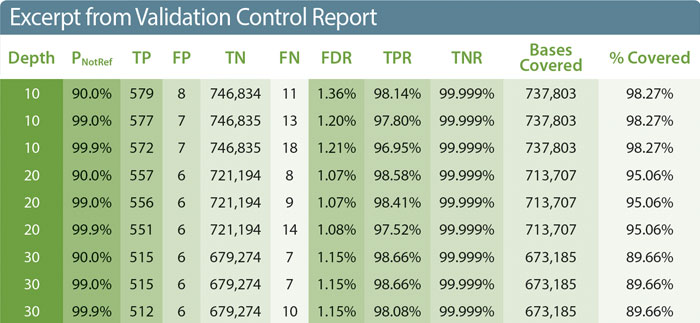
Three out of twelve depth of coverage cutoffs and three of six calculated statistics are shown. Abbreviations are as described in the text.
Conclusion
NGS-based genetic tests promise to greatly enhance patient diagnosis and care as we move into the era of personalized medicine. Given the potential impact of those tests on treatment decisions, it is critical that those tests are appropriately validated both as part of their regulatory approval and as a measure of routine assessment of the test’s performance. The NA12878 reference materials developed by NIST/GIAB together with DNASTAR’s validation control workflow greatly facilitate this process. Additional reference materials for other genomes are currently being developed and combined with new software capabilities to expand the range of tests that can be robustly validated.
At DNASTAR, Tim Durfee, Ph.D. ([email protected]), is a principal scientist, Dan Nash is a senior software engineer, Ken Dullea is a senior software engineer, Jacqueline Carville is a marketing specialist, and Frederick R. Blattner, Ph.D., is founder, president, and CEO.
At Nephropath, Marjorie Beggs, Ph.D., is the lab manager, Jon Wilson, M.D., is a nephropathologist/molecular pathologist and, Chris Larsen, M.D., is a renal pathologist.

