
March 15, 2012 (Vol. 32, No. 6)
Expanding on a concept originally coined by Walter Gilbert in 1986, Thomas Cech recently described two RNA worlds—a hypothetical, primordial world in which the same molecule combined informational and catalytic properties, and a contemporary world, forged by a spectrum of RNA-centered activities. While during the early days, following the discovery of DNA, RNA was thought to be the less interesting molecule, this view has dramatically changed in recent years.
Advances in RNA biology are reshaping concepts that have been prevailing for a long time. For example, in the field of developmental biology, decisions were traditionally envisioned mostly in terms of the progressive expression of different combinations of transcription factors that are induced by specific combinations and concentrations of growth differentiation factors.
“This is still the prevailing concept in the field, but there is much more to development than transcription factors,” says Seth Blackshaw, Ph.D., associate professor in neuroscience at the Johns Hopkins University School of Medicine.
Investigators in Dr. Blackshaw’s lab are primarily focusing on understanding the mechanisms that allow the different neuronal cell types to be specified in the adult brain during development. In a completely unbiased screen that sought to identify genes expressed at different developmental stages in the retina, Dr. Blackshaw and colleagues found several long noncoding RNAs that exhibited very prominent dynamic and cell-specific expression patterns, and their overexpression or knockdown exerted a dramatic effect on retinal development.
“We identified and studied only three long noncoding RNAs, but this is part of a bigger picture that, in terms of our notions about the genome, is in many ways comparable to the discovery of the New World,” says Dr. Blackshaw.
“There are roughly 20,000 protein-encoding genes in the vertebrate genome, and anywhere between 5,000 and 10,000 long noncoding RNAs in mice and humans, many of which are expressed in the brain, and many of which are expressed in development and, therefore, we really need to expand our notion of the real number of genes that are important molecular players.”
Understanding the biology of long noncoding RNAs is filled with challenges, and one of those is that even though a phenotype may be observed, the mechanisms of action are more elusive, making progress somewhat slow. Mechanisms are fairly straightforward for microRNAs, but more challenging for long noncoding RNAs, and it is not clear whether they work by base pairing or through completely different means.
“RNA can perform many of the same functions that proteins do. It can fold into many different shapes, it can interact with different structures, and it can even be catalytic. Almost anything that a protein can do, a long noncoding RNA can do as well, and this makes identification of the mechanisms of action of this very large set of molecules somewhat less than straightforward,” explains Dr. Blackshaw.
A recent finding from Dr. Blackshaw’s lab is the characterization of Six3OS, a long noncoding RNA that is divergently transcribed from the distal promoter of Six3, a homeodomain transcription factor important during neurodevelopment.
Dr. Blackshaw and colleagues found that Six3OS knockdown caused a dramatic phenotype overlapping with the phenotype caused by manipulating the associated protein-encoding gene, and similar defects in the development of different retinal cell types were visualized in both instances.
“We found that we could reverse the phenotype caused by Six3 overexpression or knockdown by manipulating the expression of Six3OS, and this provided conclusive evidence that these two genes interact functionally, and also suggested that the long noncoding RNA acts in a transcript-independent manner,” explains Dr. Blackshaw.
While this revealed that Six3OS regulates Six3, the mechanisms were still elusive. To address this, Dr. Blackshaw and colleagues turned to a tool that they developed in collaboration with Heng Zhu’s lab from the department of pharmacology and the Center for High-Throughput Biology at the Johns Hopkins University School of Medicine, which used Cy5-labelled Six3OS RNA to bind a human protein microarray containing approximately 70% of all annotated genes expressed as a yeast library of N-terminal GFP fusion proteins. This tool allowed the quick identification of the proteins that selectively interact with the fluorescently labeled RNA.
“We reasoned that any functionally important target of Six3OS would selectively interact with the human and the mouse homolog of the gene but not with other long noncoding RNAs,” says Dr. Blackshaw.
This completely unbiased screen identified five proteins that selectively interacted with both the human and mouse version of Six3OS, and included Eya1, a known transcriptional cofactor of Six3, along with enhancer of zeste homolog 2, a polycomb complex component that interacts with many long noncoding RNAs, and SMARCE1, involved in chromatin decondensation and transcriptional activation.
These interactions were all confirmed by immunoprecipitation in cultured human cells. This suggested that Six3OS acts not by regulating Six3 expression but, rather, that it cooperates as a specific Six3 transcriptional cofactor.
“The two are co-expressed, and the long noncoding RNA acts as a transcriptional scaffold that helps recruit transcriptional co-activators and co-repressors that allow Six3 to activate or repress its target genes, and this may be a common feature of these long noncoding RNAs, to modulate the activity of protein encoding genes that they are co-expressed with,” explains Dr. Blackshaw.
Different Approaches
“We first used hybridization-based approaches to discover a preponderance of large noncoding RNAs, then massively parallel sequencing approaches revealed their fine grain structures, and now a combined hybridization-sequencing approach being used to unravel even higher resolution transcriptional dynamics,” says John L. Rinn, assistant professor of stem cell and regenerative biology at Harvard Medical School and senior associate member at the Broad Institute.
The past few decades witnessed several alternating periods of hybridization-based or sequencing-based strategies, which culminated with their combined use. The first genetic loci were roughly mapped through crude hybridization-based methods. Yet upon the development of reliable nucleotide sequencing methods 20 years later, a dramatic shift occurred toward sequencing-based methods. That was until the sequencing of the human genome and the prospecting to determine the number of the genes within the human genome.
The solution was the emergence of DNA microarrays, which afforded tens of thousands of sequences to be simultaneously monitored. More recently, the massive parallel sequencing revolution brought sequencing back to the center stage for most applications. However, over the past few years, an approach that involves targeted hybridization followed by sequencing has been developed, and allows much deeper investigation into specific genomic fragments, such as disease susceptibility regions.
“When we were in the array era, we could find and monitor more genes by array hybridization, and now in the sequencing era, many people have shifted focus to sequencing-based gene discovery. History has witnessed these dogma shifts between hybridization and sequencing, but it is really the integration of the two approaches where one can see even more synergistic power,” explains Dr. Rinn.
An important recent advance in the field was the development of RNA CaptureSeq through a collaborative effort between the labs of Gregory Hannon, Ph.D., and Richard McCombie, Ph.D., at the Cold Spring Harbor Laboratories, together with investigators at Roche NimbleGen.
RNA CaptureSeq involves the use of tiling arrays across genomic regions of interest, followed by the subsequent hybridization, elution, and sequencing of the corresponding cDNAs. This strategy has continued to evolve to combine the strengths of in-solution capture and exome sequencing.
Recently, Dr. Rinn and colleagues applied RNA CaptureSeq to dig into the depths of the noncoding transcriptome. The authors identified previously unknown splicing patterns and isoforms, even in genes that have been extensively studied, such as p53, and in several long noncoding RNAs. Collectively, their application of hybrid-capture technology shed new insights while realizing that the full complexity of the human transcriptome is still far from being understood.
“This approach allows us to focus on long noncoding RNAs and, after capturing them, to visualize splice variations and gene-expression regulation differences in wild-type versus tumor samples,” explains Dr. Rinn.
microRNA
Approximately 98% of the transcriptional output of the human genome is represented by noncoding RNAs, which play fundamental roles in shaping the complexity of genome architecture and dynamics. MicroRNAs, a class of small noncoding RNAs that post-transcriptionally regulate gene expression, have important roles in processes that govern development, physiology, and disease. Two-thirds of the human microRNAs are located within introns of protein-encoding genes.
“For these genes, there is a unique possibility that microprocessing and splicing is coupled, either directly through protein-protein interactions or indirectly through the pre-mRNA where both macromolecular complexes assemble,” says Carl D. Novina, M.D., Ph.D., associate professor of microbiology and immunobiology at the Dana-Farber Cancer Institute and Harvard Medical School.
In a study that analyzed miR-211, a microRNA that suppresses melanoma invasion and is expressed from intron 6 of melastatin, Dr. Novina and colleagues recently showed that miR-211 microprocessing promotes splicing of exons 6 and 7, and mutations at the 5 splice site reduced miR-211 biogenesis. These findings revealed a novel physical and functional coupling between gene splicing and microRNA biogenesis and unveiled a new layer of regulation.
“In this feed-forward model, microprocessing facilitates splicing and splicing facilitates microprocessing at intronic microRNA loci. It is possible that microRNAs may play a broader role in regulating gene expression at the level of mRNA maturation in addition to their roles in regulating mRNA stability and translation,” explains Dr. Novina.
“Performing RNA sequencing works fantastically well, and it does not cost very much, but the software for analyzing the giant datasets is very challenging and still evolving, making the informatics side really hard,” says Vance Lemmon, Ph.D., professor of neurological surgery at the University of Miami School of Medicine.
Dr. Lemmon and colleagues recently used RNA-Seq to compare isoforms expressed in peripheral neurons from dorsal root ganglia, which are able to regenerate after injury, with those from cerebellar granule neurons, which do not regenerate. This comparative approach unveiled over 8,000 differentially expressed isoforms between the two cell types.
“After comparing gene expression in different types of neurons, we hope to exploit this information and transfer it from neurons that regenerate to neurons that do not regenerate, to identify targets that promote neuronal regeneration in the central nervous system,” explains Dr. Lemmon.
In addition to being very economical, RNA-Seq has several additional advantages. “This approach offers the additional opportunity to obtain information about isoforms that have never been studied and might not be in any databases,” says Dr. Lemmon. The possibility to use very small amounts of starting material also helps gain insight into the biology of defined groups of cells.
“With the possibility to identify all the RNA species from such a small amount of starting material, we can define much more precisely the specific roles they play in the different cell types from different brain regions, or under many different conditions, such as during development, disease, or injury, and this represents more information than anyone was able to get in biology before, and it is all happening in real time,” says Jessica K. Lerch, Ph.D., first author of the study.

Researchers at the University of Miami School of Medicine are using RNA-Seq to compare isoforms expressed in peripheral neurons from dorsal root ganglia, which are able to regenerate after injury, with those from cerebellar granule neurons, which do not regenerate.
Gene Expression
For a long time, the prevailing view about regulation of gene expression was that after the transcription initiation was finished, the subsequent process of elongation was not intensively regulated or controlled. “This view changed once several laboratories, including ours, had shown the existence of factors that control the rate and processivity of transcriptional elongation catalyzed by RNA polymerase II,” says Ali Shilatifard, Ph.D., investigator at the Stowers Institute for Medical Research.
An important finding that emerged relatively recently was that certain mutations in genes that regulate this process are linked to human malignancies and other diseases, elevating transcriptional elongation control to the center stage of cellular processes relevant for human development and disease pathogenesis.
“In the past eight to ten years, we have learned not only that transcriptional elongation control is central to the process of gene expression, but also that there is actually a gene class specificity such that an elongation factor could exist for every class of gene,” explains Dr. Shilatifard.
RNA biology has shaped some of the most significant moments in the history of life sciences. From the 1967 discovery that RNA can perform catalytic functions, to the seminal finding in the early 1970s that it can be copied into DNA by reverse transcriptase, and culminating with the more recent years in which considerable focus has centered on splicing and RNA interference, RNA biology has become one of the most dynamic research areas.
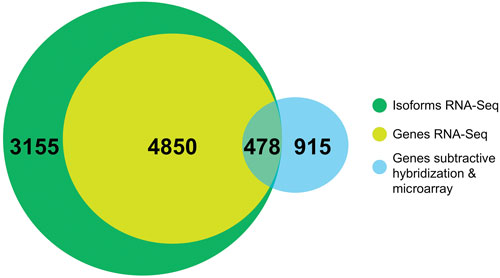
Comparison of RNA-seq to microarray or subtractive hybridization approaches to study differential gene expression. When gene-expression levels were compared between sensory neurons and cerebellar neurons many more genes were found by RNA-seq. In addition, several thousand unique isoforms were found that could not be found with the older methods. [Adapted from Lerch et al., PLoS One. (2012) 7:e30417.]
Moving RNA Devices into the Clinic
Several characteristics of RNA, which include its ability to function as a sensor and a regulator, and the ease to design it into various structures, facilitated the emergence of synthetic RNA devices that can be exploited to modulate cellular activities.
“There are multiple key ways in which RNA devices will be important in the clinic,” says Christina D. Smolke, Ph.D., assistant professor of bioengineering at Stanford University.
In addition to the lack of a requirement for heterologous proteins, which can often elicit nonspecific immunogenic responses in human cells, RNA devices are encoded within very compact platforms that do not place a large burden on the cells. One of their more immediate therapeutic applications is within cell-based therapies, such as stem cell therapy or immunotherapy.
While T-cells or stem cells used in cell-based therapies have to persist in the organism long enough to exert their specific functions, their unchecked proliferation could contribute to a different type of cancer and represents a major concern. RNA devices address this shortcoming by providing a programmable control system that precisely regulates therapeutic activities in response to different inputs.
“These inputs may be an approved, nontoxic drug molecule, or an endogenous disease signal that activates cells when they are in the specific disease microenvironment,” Dr. Smolke adds. By building a synthetic RNA device that includes an aptamer as the modular sensor and a hammerhead ribozyme as the gene-regulatory component, Dr. Smolke and colleagues recently illustrated the possibility to trigger the in vivo proliferation of T cells and modulate their growth rate in response to drug molecules.
“The cells will be engineered ex vivo to harbor genetic circuits containing RNA control devices and then ultimately introduced back into the patients,” says Dr. Smolke. The engineering of specific therapeutic activities into T cells enables investigators to regulate their proliferation and activation in the human body, allowing better temporal and spatial control and providing a safer and more effective therapy. The modularity enables the device to be easily tailored to specific therapeutic needs and represents an additional advantage.
“The proof of concept is there, and the next steps are developing integrated, robust systems, directing them toward the very relevant clinical activities that have to be regulated, and moving them into systemic animal models.”

