
September 1, 2015 (Vol. 35, No. 15)
Detecting and Analyzing Genomic Modifications Induced with Engineered Nucleases
Engineered nucleases have created unprecedented possibilities for targeted genome editing in numerous species and cell types that were once beyond the reach of targeted genome-editing approaches. These engineered nucleases include zinc finger nucleases (ZFNs); transcription activator-like effector nucleases (TALENs); and the clustered regularly interspaced short palindromic repeats (CRISPR) Type II system, which incorporates guide RNA and the CRISPR-associated (Cas) nuclease, Cas9.
Irrespective of the type of engineered nuclease used, the ultimate goal is to induce sequence-specific DNA double-strand breaks (DSBs) within the cells of interest. These DSBs can be subsequently repaired through either nonhomologous end joining (NHEJ) or homology-directed repair (HDR), resulting in the introduction of mutations into the targeted locus.
The lack of sensitive and fast methods to detect and characterize the induced genetic changes poses a major limitation to the applications of precise gene editing. This is further complicated by the fact that often only a small population of transfected cells contains the desired mutation, necessitating the screening of a multitude of clones. We have developed several methods that enable fast, sensitive, and reliable detection and analysis of indels (insertions and deletions) induced by precise gene targeting.
Direct Detection of Locus-Specific Modifications
Thermo Fisher Scientific’s GeneArt® Genomic Cleavage Detection (GCD) kit is an enzyme mismatch cleavage assay. Following the delivery of engineered nucleases into the cell of interest, the targeted locus is amplified from the extract, the genomic DNA, by means of the polymerase chain reaction (PCR) technique. The amplified DNA is then denatured and re-annealed to generate mismatched duplexes containing strands with indels annealed to strands with no indels, or strands with different indels.
The mismatched DNAs are subsequently cleaved by the detection enzyme mix, and the resultant bands are analyzed using gel electrophoresis and band densitometry. By comparing the relative proportions of cleaved and uncleaved products, one can quantitatively measure the cleavage efficiency of the engineered nuclease on the target locus (Figure 1A).
- We have optimized and streamlined the entire experimental process that can be completed within 5 hours at a relatively low cost: First, we have identified a cell lysis buffer that efficiently extracts the genomic DNA from 50,000 to as many as 2 million transfected cells with just 25 minutes of incubation (Figure 1B).
- We have optimized a PCR condition that supports efficient and reliable amplification of target genomic DNA, including GC-rich regions, directly from the crude cell lysate. In most cases, a single round of PCR amplification generates enough PCR products to perform enzyme cleavage without the need for further purification (Figure 1C).
- We have optimized the enzyme mix and reaction buffer to achieve high cleavage efficiency with low background cleavage.
- The assay allows detecting as low as 1% and up to over 90% cleavage efficiencies for various target loci and cell lines (Figure 1D). Furthermore, the cleavage detection assay is amenable to 96-well format, enabling high throughput screening and detection of targeted modification.
The GeneArt GCD assay provides a simple, reliable, and rapid method for the quantitatively detecting locus specific cleavage of genomic DNA.
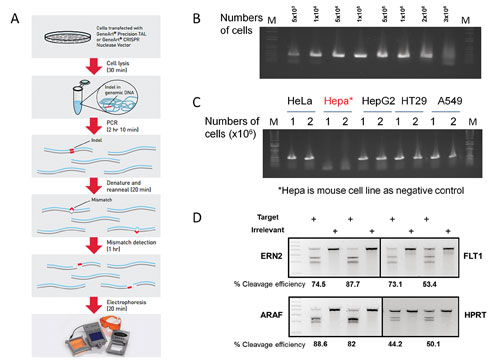
Figure 1. (A) GeneArt Genomic Cleavage Detection kit workflow. (B) PCR amplification from cell extracts lysed from various cell numbers. (C) PCR amplification using genomic DNA extracts of different cell types. (D) Genomic cleavage analysis of samples with CRISPR gRNA-Cas9-transduced cells.
Rapid Screening of Cleavage Activity and Enrichment
To provide a fast screening tool for cleavage activity of gene-editing tools, we have developed the GeneArt Genomic Cleavage Selection (GCS) kit. The design of the GeneArt GCS vector is based on the observation that when a target sequence on the chromosome is mutated by the engineered endonucleases, the same target sequence on a plasmid in the same cell is also likely to be mutated. The GeneArt GCS vector contains two tandem copies of the partial overlap coding sequence of the orange fluorescent protein (OFP) reporter, which are separated by the insertion of the target sequence of the programmable nucleases.
When cells are co-transfected with the reporter construct and engineered nucleases, a double-stranded break is introduced into the target sequence by the nuclease. The two repeated OFP sequences can then recombine to yield a contiguous OFP coding sequence, thus restoring OFP and CD4 reporter gene expression (Figures 2A & 2B).
When this reporter vector is used, it is possible to verify cleavage activity of any engineered nuclease system, including ZFN, TALEN, and CRISPR/Cas9, as early as 24 hours post-transfection by simply checking for OFP expression of the transfected cells using a fluorescent microscope. The cells can be further analyzed and quantified using flow cytometry (Figure 2C). The percentage of OFP-positive cells is proportional to the cleavage activity of engineered nucleases.
In addition, expression of OFP and CD4 reporters allows for enrichment of the nuclease-modified cells from the mixed population (Figure 2D, lanes 1–4). To demonstrate this capability, we first diluted the transfected cells with parental cells to obtain cell population with a low proportion of gene modification events (lane 1). Then we obtained a flow-through sample after CD4 enrichment (lane 2). This was followed by enrichment using a cell sorter for OFP-positive cells (lane 3) or using CD4 antibody-conjugated Dynabeads® to enrich CD4-positive cells (lane 4).
The nuclease-modified cells were enriched over 20-fold based on cleavage efficiencies measured by GCD assay. This enrichment will significantly reduce the downstream workload for clonal isolation of edited cells, especially when only a small fraction of a targeted cell population is edited.
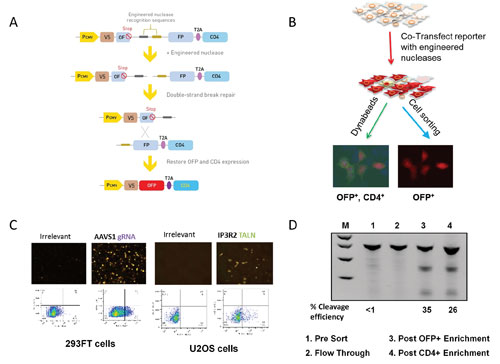
Figure 2. (A) Principle of GeneArt Genomic Cleavage Selection vector during genomic cleavage selection. (B) Workflow used with the Genomic Cleavage Selection vector. (C) Fluorescent images and fluorescence-activated cell sorting analysis of cells transfected with the Genomic Cleavage Selection vector. (D) Enrichment of nucleases-modified cells using the Genomic Cleavage Selection vector.
High-Throughput Characterization of Gene-Targeting Events
We have established a streamlined workflow for next-generation sequencing (NGS) using the Ion PGM™ System, which enables processing of up to 96 samples in one sequencing run.
Briefly, a library is generated from a 200-400 bp amplicon containing the genomic target region using the Ion Plus Fragment Library Kit. Amplicons from multiple samples can be barcoded using Ion Xpress™ Barcode Adapters and pooled into a single library, followed by amplification onto Ion Sphere Particles using the Ion OneTouch™ 2 System.
The enriched monoclonal amplicon library is then sequenced using Ion PGM System. The reads are aligned to the corresponding reference sequences and subsequently characterized and counted using Integrative Genomics Viewer or Tablet.
The resulting sequence yields thousands to millions of reads per sample, which allows users to detect a very low modification frequency (
We have compared gene-editing efficiency detected by GCS, GCD, and NGS (Figure 3). CHO cells were co-transfected with the genomic cleavage selection reporter and three pairs of PerfectMatch TAL nucleases using the Neon™ transfection system. Forty-eight hours post transfection, the percentage of OFP-positive cells was analyzed by a flow cytometer.
Extracted genomic DNA from an aliquot of the transfected cells was used as template to amplify target loci for GCD assays and NGS analysis (Figure 3A). It was determined that the percentages of OFP-positive cells detected by the GCS assays are slightly lower than in the other assays because in this analysis the OFP intensities of the transfected cells did not enter the calculation (Figure 3B). However, the cleavage efficiencies detected by GCD were in concordance with gene-modification efficiencies detected by NGS.
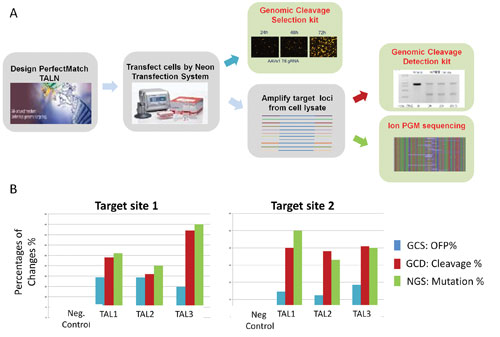
Figure 3. (A) Experimental processes for detection and analysis of targeted gene modifications. (B) Comparison of results from Genomic Cleavage Selection (GCS), Genomic Cleavage Detection (GCD), and next-generation sequencing (NGS) detection.
Discussion
The methods introduced here provide distinct yet complementary solutions for effectively detecting, characterizing, and enriching genetically modified cells. These genome-editing tools both enable and simplify a broad range of genome-editing and cell-engineering applications, including the development of gene knock-in, gene knockout, and transgenic organisms.
Jian-Ping Yang, Ph.D. ([email protected]), is a staff scientist, Natasha Roark is a scientist II, Pei-Zhong Tang, Ph.D., is a senior staff scientist, Veronica Blackston is a scientist II, and Jonathan D. Chesnut, Ph.D., is a senior director in R&D at Thermo Fisher Scientific.

