
June 1, 2012 (Vol. 32, No. 11)
Chris Torrance, Ph.D.
New Genomics-Focused Technologies Can Help Alleviate Points of Historical Attrition
These are interesting times to be in the drug discovery field. The blockbuster business model is falling by the wayside, driven in large part by rapid advances in our understanding of the myriad of molecular drivers of cancer and other complex diseases, rising efficacy bars for new targeted or personalized therapies, and the adoption of value-based (sometimes zero) reimbursement practices for marginally effective drugs or treatment regimens that continue to gain approval.
The increasing move toward addressing rational molecular targets that directly drive disease biology and biomarker-driven clinical practices that enable the tailoring of novel drugs to only the patients that harbor the given molecular target is of benefit to patients and society as both maximally effective outcomes, and management of future healthcare costs will be ensured.
This trend, however, also places extreme pressure on pharmaceutical companies to reduce their timelines, attrition rates, and costs in order to find many more targeted drugs for smaller but more responsive patient populations. There is also an imperative for cancer biologists and the biotech sector to decipher the full genetic complexity and heterogeneity of cancer. This will define next-generation targets for drugging and clinical scenarios in which they have the best shot for gaining approval.
The genomic landscape of cancer (and many other diseases) will represent ground-zero data for the majority of future drug discovery efforts. These are the clearest indicator of what is potentially important to a tumor; and with DNA sequencing no longer representing a bottleneck, the full range of genetic mutations, their frequency within various tumor types, and their staging information will soon be known.
This is being driven by the coordinated efforts of multiple global genome centers, which are making all data publically available. These efforts will in the future deliver more complex and useful datasets, such as correlating which cancer genes commonly occur together (so that candidate drug response–defining biomarkers and potential drug combination strategies can be defined) or which mutations represent early events (to identify targets with less intratumoral heterogeneity). Recapitulating this sequencing and bioinformatics infrastructure within any pharma company will be pointless; however, deciphering which cancer genes represent good drug targets will be essential.
The first step in determining which mutant genes represent good drug targets requires the definition of which genes are actually true disease drivers, rather than just random noise in genetically unstable tumors. If a gene is mutated frequently, then it is usually safe to assume that it is, or was, a driver (confirmed through validation experiments). However, most candidate cancer genes are not very common, requiring a systematic functional genomics effort to assess exactly which elicit a significant tumorigenic effect.
This would involve implementing a large, coordinated study of all the hallmarks of cancer (as not all genes will increase cell proliferation) and a new generation of genome-editing technologies to directly alter the sequence of endogenous genes in human cells growing in cell culture.
A technique pioneered by Horizon Discovery, GENESIS™, allows the full range of genetic alterations to be made in human cells (small or large gene deletions, point-mutations, reversion of mutations to wild type, translocations, amplifications, and transgene insertions), with unparalleled precision (Figure 1). The technique harnesses the power of recombinant adeno-associated viruses to activate homologous recombination, a natural high-fidelity DNA repair mechanism.
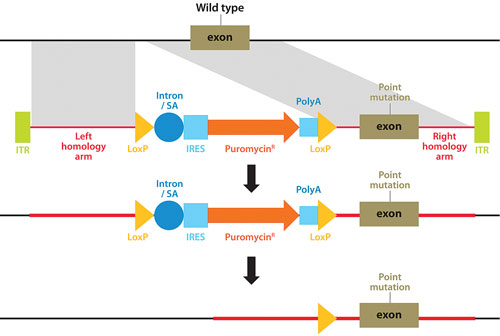
Figure 1. Genome editing using rAAV-mediated homologous recombination to introduce any desired genetic alteration into a target endogenous gene locus in human cells growing in culture.
Disease Models
A natural extension of the genome editing approach, and one that will be a key component of enabling high-throughput and systematic target identification and confirmation, is the ability to recreate patient genetics in the laboratory. Although cell lines harboring common cancer genes are available for research, less common genes are poorly represented or often cannot be sourced.
Endogenous target genes would also need to be modified with cutting-edge pathway reporter tools (e.g., Halo-Tag™) so that these rarer cancer genes can be functionally assembled into larger druggable networks and then applied in direct screens for novel targets or even drug candidates.
Panels of unrelated cell lines differ genetically in thousands of ways, making deciphering gene or drug function without an initial hypothesis a difficult process. This is where “isogenic” disease models—cell-line pairs that share the same genetic background except at a specific locus modified by gene editing—will be invaluable to definitively study disease biology and profile candidate drugs against specific biomarkers through the entire discovery process (Figure 2).
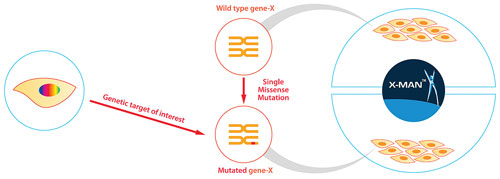
Figure 2. Genetically defined and isogenic cell-line panels, created by GENESIS, that are perfectly matched and harbor a specific normal vs. mutant patient-relevant genetic feature.
Target Validation
It is crucial to validate new targets that possess robust disease dependencies, which if chosen wisely at the outset can yield faster moving programs than historically achievable (Figure 3). An inability to consistently validate, coupled with difficulty in progressing, or cancelling, lead discovery programs at logical and early decision points, has been a primary source of high attrition rates over the last two decades.
Moving forward, we also face the immediate inconvenient truth that the majority of the cancer genome is not directly druggable at this time (e.g., nonenzymatic oncogenes or physically absent tumor suppressors) and thus “deorphaning” the cancer genome will now become a de facto step in the discovery process.
One solution is to combine RNAi-based screens with genetically defined disease models, enabling the systematic isolation of targets with penetrant and selective antitumor effects: holistically accessing all aspects of tumor biology, including unpredictable “synthetic lethalities” that can occur as cancers rewire their cellular networks. Once strong candidate targets have been isolated, these can be further validated again using genome editing to create, for example, genetic knock-outs, silent mutations that rescue RNAi effects, or even subtle activity-dead versions to mimic the action of a small molecule inhibitor.
The final step in any personalized drug discovery toolbox will be to enable the prediction (or confirmation) of which patients are likely to benefit, so that small, rationally selected clinical trials can be performed. In vitro disease models (and their xenografts) will certainly play their part again, but models that access more complex tumor-stroma interactions, such as patient-derived explants and genetically engineered mouse models, will also be important to confirm that drugs can function in a tumor-relevant context.
With a rapidly changing environment, all stakeholders in the drug discovery continuum now have an opportunity to come together to accelerate the discovery of personalized medicines. Transformative genomics-focused technologies are now available that will alleviate points of historical attrition: from choosing more robust targets, to better on-target drugs, and, finally, finding the right patients to enable faster drug development.
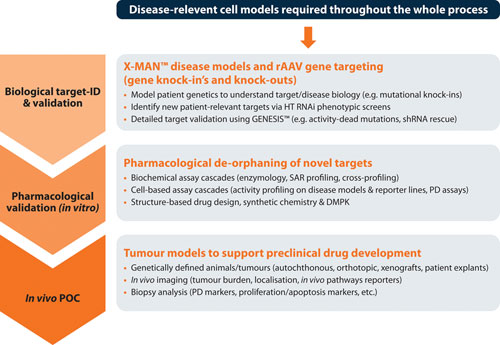
Figure 3. The target validation toolbox, with genome editing and disease models supporting each stage of drug development with clear on-target, patient-relevant data.
Chris Torrance, Ph.D. ([email protected]), is CSO at Horizon Discovery.





