July 1, 2012 (Vol. 32, No. 13)
Next-generation sequencing (NGS) allows the interrogation of genomes and transcriptomes at unparalleled resolution. NGS is becoming a powerful tool to identify cancer mutations that will eventually be translated to the clinic.
Further, second-generation RNA-Seq technology permits the simultaneous evaluation of gene expression and transcript structure at a high level of accuracy and at a single-nucleotide level. RNA-Seq has been called a revolutionary tool for transcriptomics. It works by utilizing NGS high-throughput technology to characterize cDNAs representative of the cell’s transcriptome.
RNA-Seq can be a valuable analytical tool for a variety of applications, notes Erik K. Flemington, Ph.D., professor of pathology, Tulane Health Sciences Center. “In my laboratory, we have utilized this technology to identify and analyze the transcriptomes of infectious viral organisms, to characterize tumor microbiomes, and for microRNA (miRNA) target analysis studies.”
Dr. Flemington says that often viruses have a high gene density, making it difficult to discriminate overlapping transcripts using RNA-Seq. “Newer RNA-Seq methodologies are allowing us to overcome those challenges. Using these methods, we are finding out that the old dogma that there are only a small number of transcripts isn’t true. In fact, we have identified an abundance of previously unannotated and/or undescribed transcripts in viromes.”
As an example, Dr. Flemington and colleagues studied Epstein-Barr virus (EBV), a human pathogen that causes malignancies such as Burkitt lymphoma and Hodgkin disease. “We used second-generation RNA-Seq pipeline tools and developed new tools to customize the approaches for the analysis of viromes in the context of their host.
“Among other things, these new strategies allowed for the identification of new viral genes and transcript isoforms important for EBV to establish infection. Overall, these studies allowed us to identify a whole new set of transcripts that are potentially related to such processes as cell fate determination and inflammatory events.”
Another use of RNA-Seq is to characterize tumor microbiomes. “Clinical samples may contain exogenous agents such as viruses. This is important to know because some of these contribute to tumor development. By assessing clinical samples with RNA-Seq we can discover if the tumor has viruses associated with it.
“An example is the analysis of stomach cancers. The identification of viruses in clinical samples is highly tractable, and instead of needing to perform numerous assays looking for each virus one at a time, RNA-Seq allows identification in one assay alone. As this technology is used more and more in clinical samples, we may be able to better determine which viruses are associated with which tumors and what the clinical significance of these interactions is.”
Dr. Flemington also employs RNA-Seq for miRNA-targeting studies. “The regulation of gene expression by miRNAs is a fundamental mechanism for controlling a number of biological processes. We used RNA-Seq to study, for example, miRNA-155. The gene encoding miRNA-155 was classified as an oncogene long before it was identified as an miRNA. It is now implicated in a wide variety of cancers.
“Previous studies have utilized microarrays to assess miRNA-mediated decreases in target RNA. But this approach suffers from technical limitations. We employed RNA-Seq because of its high level of accuracy, broad dynamic range, ability to assess transcript structure, and because it can sensitively assess transcriptome alterations. Using this approach, we were able to identify a large inferred targetome, and more interestingly, we could readily study the role that transcript structure plays in microRNA targeting.”
RNA sequencing of individual circulating tumor cells can be used to detect the generation of drug-resistant clones in blood from patients undergoing breast cancer therapy. [National Cancer Institute]
Integrative Analysis
The ability of RNA-Seq to generate millions of reads has presented new challenges to data analysis and interpretation, notes Han Liang, Ph.D., assistant professor, department of bioinformatics and computational biology, University of Texas MD Anderson Cancer Center. “We are studying the molecular underpinnings of gastric cancer using an RNA-Seq approach. The huge amount of data generated required us to develop creative in-house ways to interpret and analyze it.”
In a recent study, Dr. Liang and colleagues profiled the transcriptomes of gastric tumor and noncancerous samples from the Asian population. “Gastric cancer is the most common cancer in developing countries and the second leading cause of cancer death in the world.
“Traditional approaches to study gastric cancer have utilized hybridization microarrays, including miRNA expression microarrays and exon microarrays, but those approaches only characterize some part of the transcriptomes. We chose RNA-Seq to perform this analysis and generated 680 million informative short reads of these transcriptomes. This included profiling mRNA and miRNA simultaneously.”
Dr. Liang and collaborators applied a SOLiD™ RNA-Seq (Life Technologies) approach and developed a multilayer and integrative approach for characterization. “We utilized two complementary protocols for generation of a target fragment library that ranged from 50–150 nucleotides in length as well as shorter reads from 18–40 nucleotides. In this way we generated reads on the entire population of transcribed molecules.”
The next challenge was to analyze the data. “We performed a multilayer and integrative analysis on the data and identified different types of transcriptional aberrations that were associated with different stages of gastric cancer. We used a combination of commercially available software and our own in-house algorithms. In order to integrate expression data of mRNA and miRNA, we developed algorithms to quantify and compare gene-expression patterns.”
Their analyses pinpointed a potentially functional target. “We identified the central metabolic regulator AMP-activated protein kinase (AMPK)α2. Thus, this gene is a potential therapeutic target for early-stage gastric cancer in Asian patients.”
Dr. Liang plans to utilize this system and NGS for gastric cancer patients in other populations. “Ultimately, we hope to identify the most important biomarkers in gastric cancer. Using RNA-Seq we can start with a more global dataset and then narrow that down in each population, eventually in each patient.”
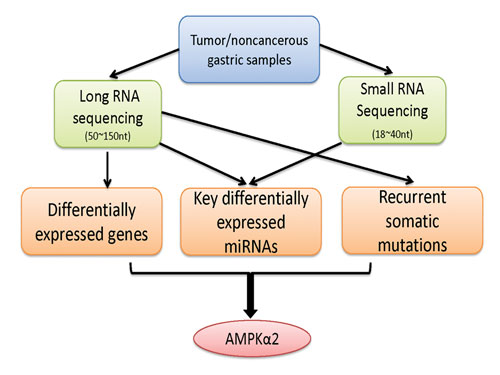
A multilayer and integrative analysis of the whole transcriptome in gastric cancer: Tumor and noncancerous samples were first subjected to two complementary sequencing protocols that target the RNA fragments from 50–150 nucleotides and 18–40 nucleotides, respectively. Then a multilayer analysis was performed to identify different types of transcriptional aberrations that were associated with gastric cancer, including differentially expressed mRNAs, key differentially expressed miRNAs, and recurrent somatic mutation candidates. Finally, the integrative analysis suggests AMPKa2 as a potential functional target in Asian gastric cancer. [University of Texas MD Anderson Cancer Center]
Single-Cell Analysis
The sensitivity and precision afforded by RNA-Seq makes it an ideal technology to characterize the gene-expression signatures of circulating tumor cells (CTCs). “CTCs are often used for early diagnosis and monitoring of responses to cancer therapies,” notes Abizar Lakdawalla, Ph.D., marketing manager, new technologies, Illumina.
“The gene-expression profile is usually unique to the cell’s lineage (where the CTC has originated). It clearly shows the level of heterogeneity in a patient if multiple CTCs are isolated and sequenced from that individual’s blood. Further, RNA sequencing shows changes in RNA structure (different isoforms formed by alternative splicing, novel RNAs produced by translocation or other structural changes in the genome) that often correlate with the origin and progression of a disease.”
Dr. Lakdawalla says that RNA sequencing of individual CTCs also can be extremely useful for detecting the generation of drug-resistant clones from blood for individuals undergoing therapy. “This allows for the optimization of the therapy regimen with the type of lesions prevalent in new CTC clones.”
In order to perform single-cell sequencing, a number of modifications in the procedure needed to be optimized. Dr. Lakdawalla notes, “RNA sequencing of individual cells is based on three steps. First, individual CTCs are isolated from blood by immunolabeling them with cell-specific markers and isolating them through magnetic affinity capture.
“The mRNA in the single cell is converted to cDNA from which a sequencing library is derived with a modified Illumina library prep method. This is quantitated and sequenced on a sequencer with paired 50 or 75 bp reads.”
There are a number of potential applications for RNA-Seq of individual cells, according to Dr. Lakdawalla. “The method is applicable to samples where there is a limited amount of biological materials. Molecular phenotyping of CTCs from blood, single-cell capture, and RNA sequencing is extremely useful for the isolation of CTCs or other cells from breast exudates (breast cancer), urine (kidney-related diseases), and feces (colorectal cancers), etc.
“The high sensitivity and specificity of this method expands the range of samples that can be analyzed. For example, cells isolated by laser capture from frozen or FFPE tissues sections also can be sequenced.”
Dr. Lakdawalla also reports that single-cell RNA sequencing is highly useful in developmental biology (to track spatial and temporal gene-expression changes in individual cells in a developing embryo), to create a three-dimensional gene-expression map of an organ at millimeter resolution, and for evaluating clonal heterogeneity in production cell lines, for example.
Translation to Clinic
Utilizing targeted sequencing to identify cancer biomarkers may soon translate into better clinical patient care. “For biomarker discovery, one wants the most comprehensive way to analyze the genome,” says Olivier Harismendy, Ph.D., assistant professor of pediatrics at the Moores Cancer Center, University of California, San Diego.
“The sequencing of all exons offers great opportunities to discover cancer somatic mutations and DNA-based biomarkers that can translate into new therapies. This is a very powerful way to discover new tumor suppressor genes and oncogenes.”
There are, however, significant drawbacks to such a broad approach, which hinders its wide adoption for clinical care. First, the clinical significance of the vast majority of identified mutations is still unclear. Then, the quality of the samples collected in the clinic is suboptimal due to cellular heterogeneity.
Heterogeneity arises in several ways: inclusion of normal cells during cancer resection or biopsy and also from the presence of several subclones in the tumors themselves. In both cases it impairs the ability to detect the somatic mutations. Dr. Harismendy and colleagues have developed a streamlined approach for massively parallel sequencing of cancer mutational hotspots in heterogeneous samples.
“We devised a novel ultra-deep targeted sequencing (UDT-Seq) assay that enhances laboratory workflow and mutation detection. The idea is that targeted sequencing assesses all clinically actionable genes and allows for high sequencing coverage depth. As a result there is a much more sensitive analysis of heterogeneous clinical samples, and that enhances its clinical utility.”
The UDT-Seq is a direct sequencing assay with ~200-nucleotide long PCR amplicons generated via multiplexing utilizing microdroplet PCR.
“We initially focused on 71 kilobases of mutational hotspots in 42 cancer genes. We use chimeric primer pairs with both locus-specific and adapter sequences to generate PCR amplicons. These were directly sequenced on the Illumina Genome Analyzer II platform. This process simplifies the workflow because it removes the time-consuming and error-prone step of sample fragmentation and library preparation.”
Dr. Harismendy and team are now engaged in a pilot clinical study that uses an updated version of this assay using a faster and more accurate DNA sequencer (MiSeq from Illumina), for evaluation of 47 genes in 40 patients with breast cancer.
“The goal of the clinical trial is to improve clinical care. Somatic mutations in the tumor DNA will inform us if particular patients are eligible for a targeted treatment. Pharmacogenomic markers in the patient’s germline DNA can help avoid adverse effects of some therapies.
“Finally, some inherited DNA variants will be helpful for prognosis but could also help the patients’ relatives that could be at increased risk for cancer. We will report the validated results back to the patient’s physician. As a result we hope that some patients may be eligible to enroll in the latest targeted therapies clinical trials for breast cancer.”

