
July 1, 2017 (Vol. 37, No. 13)
Building Processes for the Future
The successes seen in a number of clinical studies on viral vector-based gene therapies (AAV, retroviral, and lentiviral vectors) are well documented, with an ever-broadening pipeline of products entering late-phase clinical trials and hopefully, through to becoming licensed medicines. Almost all of the AAV-based therapies under development target devastating and potentially fatal diseases, which severely impact the quality of life of the patient and their families (Table).
While for some diseases there may be existing treatments available, these are often very expensive and far from ideal from a patient perspective. For this reason, clinical development programs for innovative gene therapies have been actively accelerated by regulatory bodies such as the MHRA and the FDA by the application of a “Fast Track” or “Breakthrough” designation, enabling expedited access to these new medicines.
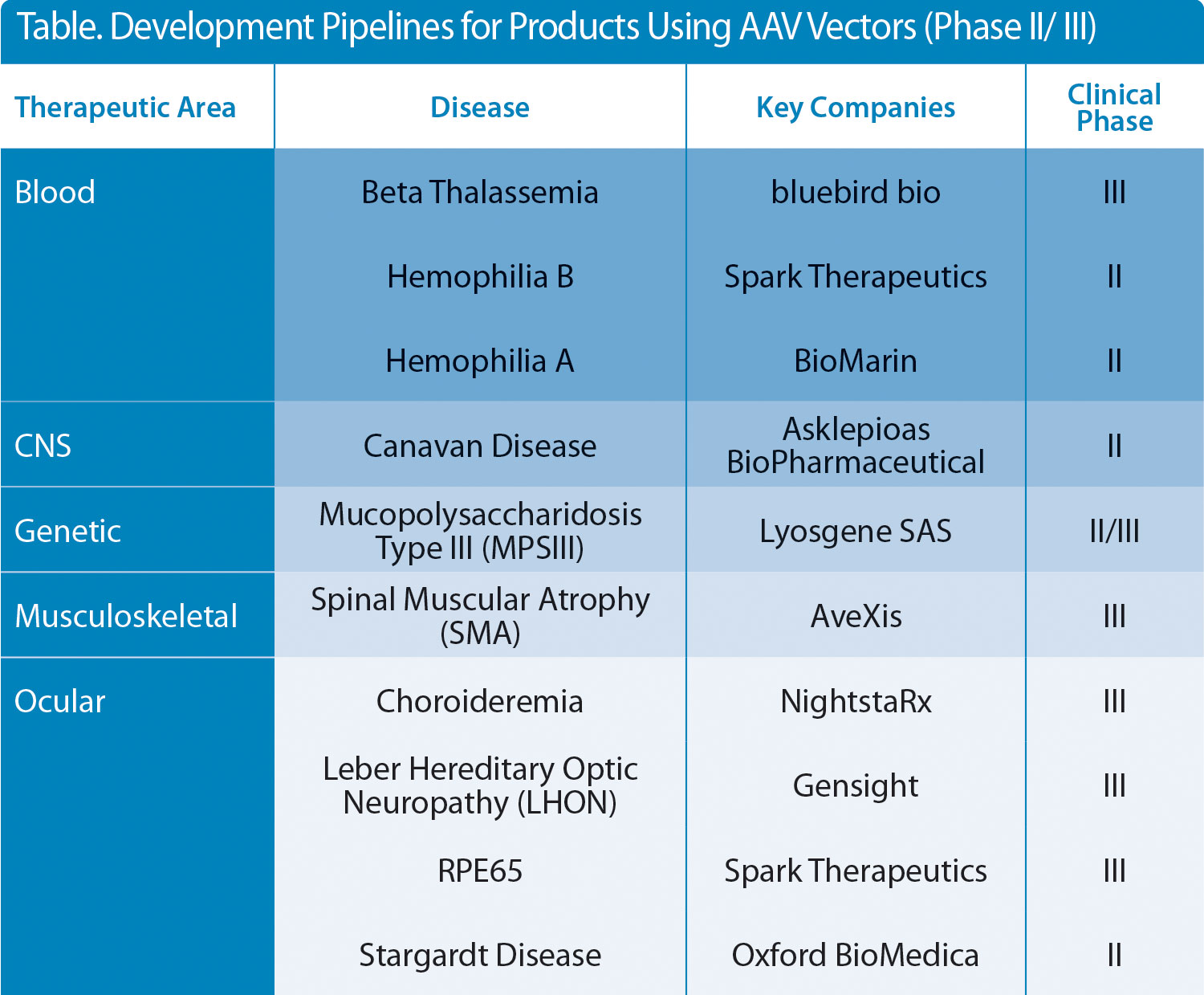
Table. Development Pipelines for Products Using AAV Vectors (Phase II/ III)
The flip side of accelerated pathways is that timelines for the manufacture of viral vectors becomes compressed, making it more difficult to develop commercial manufacturing processes that can produce vectors at the right quality, in the required amounts, and at costs that are reasonable enough to secure reimbursement from healthcare providers while still making financial sense to the developer. Although there are a number of potential approaches for the production of viral vectors, including baculovirus expression systems and the development of producer cell lines, the most commonly applied approach used to date is transient transfection in adherent cell culture systems. This approach is ideal for early-stage development in terms of speed, simplicity, and limited process development or infrastructure investment, and has been used for the majority of ongoing clinical studies (Figure 1). However, this method is recognized to have serious constraints with regards to productivity levels, process scale up, and large-scale production.
From a production and regulatory perspective, manufacturers are now faced with the dilemma of how we should approach the transition from clinical to commercial manufacturing for these highly complex products. Ideally, one would like to start from a clean sheet and rebuild the process around scalable platforms. However, time, cost, and most importantly, regulatory constraints prevent this approach.
For many products, process development opportunities are limited, with manufacturing groups having to maintain the supply of products for ongoing clinical studies, while simultaneously working to develop and improve processes. If successfully licensed and launched, it is likely that these products will be made using processes that are far from optimal. The challenge, from a manufacturing perspective is, therefore, to ensure that this approach does not restrict patient access or safety.
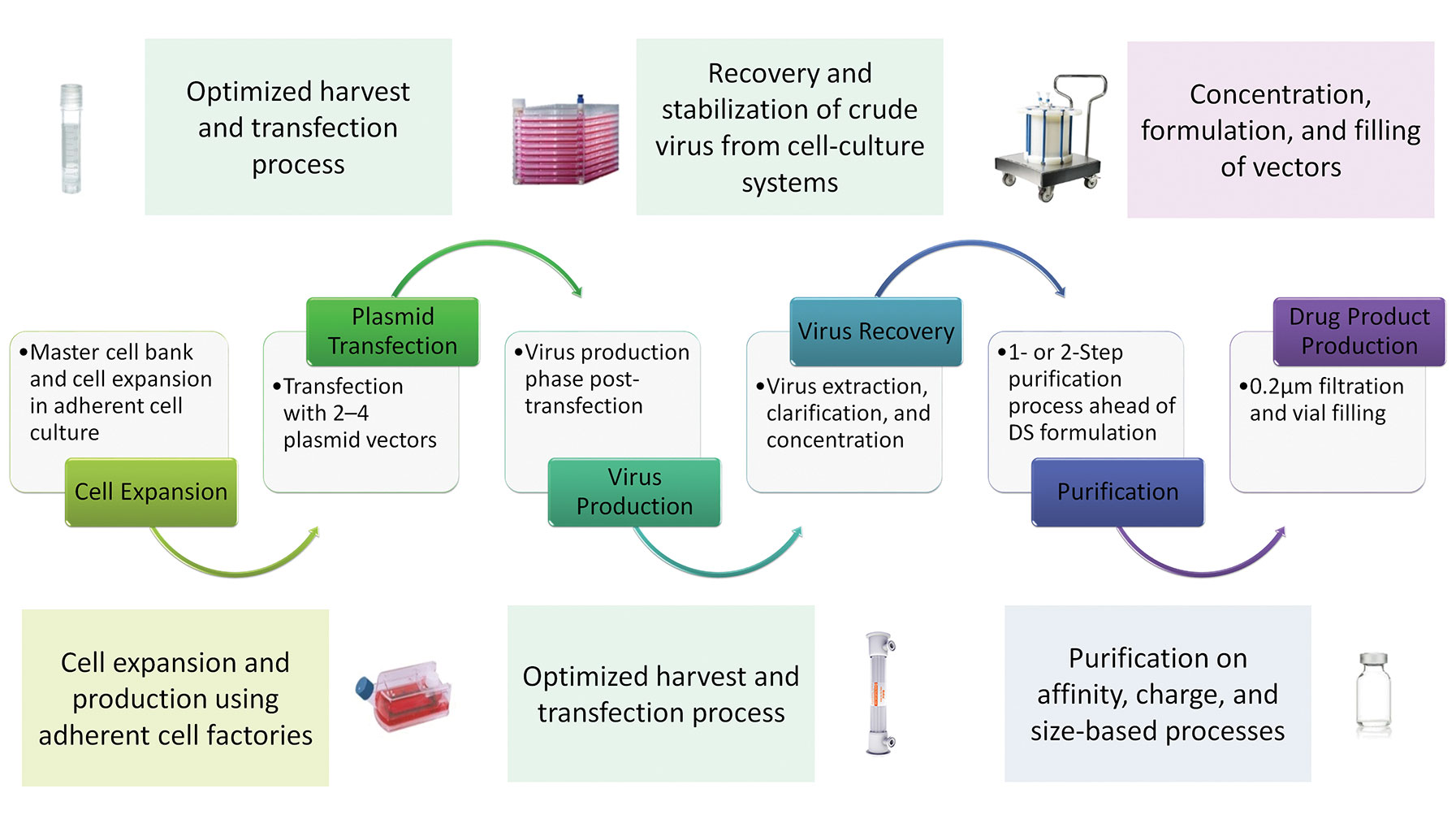
Figure 1. A sample manufacturing process using AAV vectors. DS = drug substance.
Inherent Complexity
The inherent complexity of viral vector-based products, due to their physical size, formulation, and the fact that they often utilize a combined drug targeting/delivery vehicle function, makes their physical and biological characterization highly challenging from a regulatory perspective. Consequently, a fallback approach is adopted where the product is defined by the manufacturing process. This approach then makes the introduction of potentially product-impacting process changes difficult to implement and by default, the process becomes “locked down” within the early stages of development, severely restricting the scope for process improvement and scale up.
Classical process scale up tends to be via a vertical approach, with a focus on increasing the size of single operations (such as fermentation vessels) while keeping similar labor levels, subsequently achieving reduction in cost. This approach is valid if the process is well understood and amenable to linear scale up. The reality is that a large number of the key operations in the production of viral vectors are neither well characterized nor easily scaled. Lack of time and analytical tools will eventually direct developers to take a more horizontal approach to process scale up.
It seems likely that scale up will be based on limited vertical scale up, with multiple and overlapping production streams, potentially exploiting options around the adoption of closed single-use production systems to maximize outputs from production facilities. While this may not be the most efficient approach with regard to labor and facility costs and end-product testing, it is likely to be the only realistic option for many product development groups.
It is inevitable that some process changes will need to be introduced, for example, the requirement to replace purification of vectors by ultracentrifugation, as these processes are perceived as not only being unscalable, but also as highly operator-dependent with regard to yield and purity. The challenge becomes how engineers replace this type of operation. From a regulatory perspective, the key is an understanding of the critical quality attributes (CQAs) that impact product safety, purity, and potency; the critical process parameters (CPPs) required to control them; and the availability of the tools to measure CPPs.
This approach then, in theory, will allow process development groups to develop strategies for introducing and verifying the impact of desired process changes. However, the successful process development of these “legacy” processes will be dependent on the availability of suitable in-process and final-product assays. There is a clear regulatory, as well as operational, need for drug developers to invest in the analytical tools required to achieve greater understanding of AAV vectors and the processes used to make them for the products to receive commercial licensing.
The production of vectors through transient production routes entails a complex materials supply chain. At the front end is the supply of plasmid DNA constructs used to generate the vectors; clearly the quantities required will not only increase proportionally with the increased scale of vector manufacturing, but also, the associated quality requirements will be increased, moving from materials made to “traceable” standards to those made to GMP-grade standards (Figure 2). For early-phase development, non-GMP-grade plasmids may be used for the production of material for proof-of principle clinical studies. However, this may not be the case for commercial vectors, where GMP-grade plasmids may be required. One consequence of this will be the potential need for manufacturers to align with suppliers that have large-scale GMP capabilities to ensure the timely and secure delivery of plasmid supplies to support late clinical and commercial production.
At the end of the supply chain is the production of the viral vector drug product. For early-stage development, relatively little focus is given to either the product formulation or the filling process. There is often good reason for this, as material for such development studies is in very short supply, with all available material often directed into clinical studies to demonstrate product efficacy.
The result of this is that the basic formulations used in early-stage development are carried forward into late-stage trials, with the products 0.2-µm filtered and hand filled into glass vials and stored at –80°C.
Future development activities in the AAV field will need to be focused on identifying formulations that provide long-term stability, potentially moving to +2–8°C storage, and generating meaningful stability data. Fully defining the drug product manufacturing process will also ensure the retention of product titers and activity throughout the manufacturing process, including activities such as inspection and labeling.
In conclusion, we are in exciting times with a number of these potentially life-changing products coming through to clinic. However, if we are to bring these products efficiently to the market, developers will need to adopt pragmatic and informed solutions for the manufacturing challenges that lie ahead.
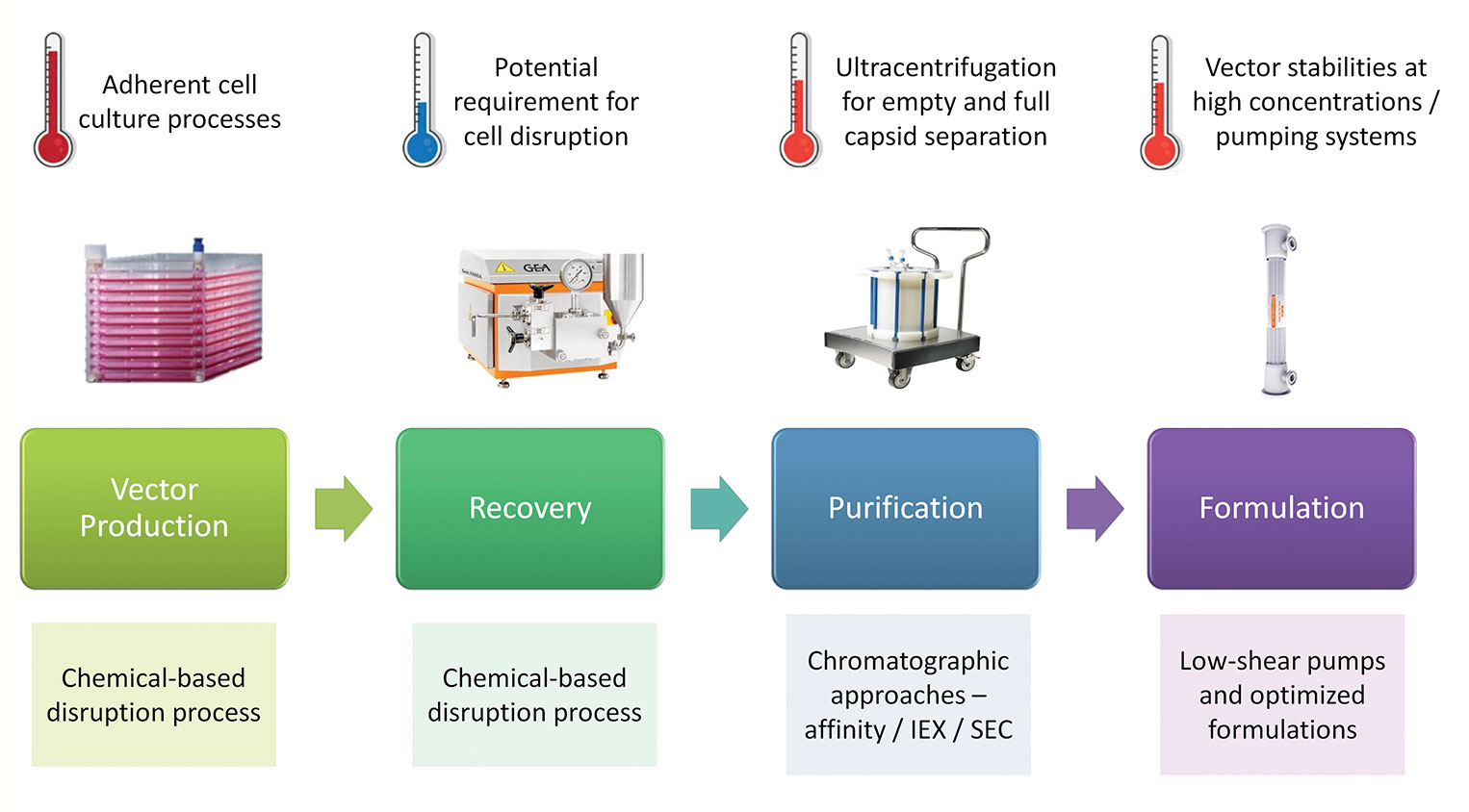
Figure 2. Challenges for scale up.

