
March 1, 2012 (Vol. 32, No. 5)
Intimate Connectivity Must Be Maintained Throughout the Product’s Lifecycle
The link between product specifications and analytical methods throughout a product’s lifecycle makes intuitive sense. One cannot change without affecting the other.
Process development groups often revise product specifications, or apply methods to an application for which it was not originally intended. A method may have originally been developed as a limits test, but now is required to assess yield.
The analytical development group must now implement modifications quickly, which becomes more difficult when analytical does not communicate directly with the processing group, as is typical at CROs.
A product specification is a list of tests, references to analytical procedures, and associated acceptance criteria consisting of numerical limits, ranges, or other standards for the tests described. Similarly a critical quality attribute (CQA) is a property either demonstrated through clinical trials, or presumed to be related to clinical safety or efficacy.
Each product specification has three key elements: target quality attributes confirmed through some analytical test, analytical test method for a particular attribute, and acceptance criteria.
Justifying product specifications depends on diverse inputs, including a method’s capabilities, regulations, process capabilities (including variation), characterization studies, clinical and nonclinical studies, and literature research.
Literature-based knowledge is often completely absent in early Phase I, yet analytical groups must nevertheless set specifications, often based on experience with similar molecules.
Perhaps the most consequential specification input is analytical method capability. The attribute of interest must, after all, be assessable. Thus specifications depend on the method’s specificity, sensitivity, variability, and accuracy.
Sensitivity and specificity relate to the ability to discriminate between a target analyte and its matrix, to unequivocally assess that analyte, and to measure it at appropriate levels.
Related are assay variability and accuracy. A product specification of 98% to 102% will not work with an intermediate precision of 10%. Contributions to variability and bias arise from instrumentation, sample preparation, the analyst, and the method itself.
Failure to account for analytical method capability may result in specifications that are unachievable, and ultimately the inability to release product. Analytical groups encountering these problems sometimes attempt to solve them by replicating experiments.
But adding replicates strains resources, delays projects, and results in avoidable cost overruns. Therefore, ongoing evaluation of the method is critical for maintaining the link between analytical method and specification.
Traditionally, the analytical group qualifies methods early in development to assure its suitability. Qualification typically reduces to a check-box exercise confirming the appropriate precision and accuracy. But a great deal occurs between Phases I and II: How many analytical scientists can guarantee that a method remains valid when its test article, environment, or intended use changes? The take-home lesson is always to consider analytical methods when revising or establishing new product specifications.
With the link between methods and associated specifications firmly established, the task becomes maintaining this connection throughout the product’s lifecycle. This may be achieved through QbD for analytical method lifecycle management.
In recent years global regulatory bodies have been urging pharmaceutical companies to adopt QbD, which the ICH defines as “a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.” Applying QbD to analytical methods gives rise to three essential elements: analytical target profile, design space, and analytical control strategy.
Analytical target profile establishes the conditions or criteria that need to be met when assessing CQAs. Examples include the ability to evaluate the CQA, select the target analyte from its matrix, and satisfy the requirements of downstream functions like process development or manufacturing.
Design space is the multidimensional combination and interaction of input variables and process parameters that demonstrate quality. When considering design space with regard to analytical methods, one should consider sources and magnitudes of variability, which in turn are associated with the ruggedness of a method with respect to the analyst, instrument, or location, and the method’s robustness.
Control Strategy is a planned set of controls, determined from product and process understanding that ensures process and product quality.
The Figure illustrates the interdependence of process control, analytical control, and product specifications. The information from the process control strategy feeds into analytical control strategy and product specifications, resulting in an environment of continuous improvement. Additionally, a constant dialogue is maintained between process development and analytical, thus maintaining the link between specifications and methods.
QbD for analytical methods can be summarized in four key steps: method performance requirements, method development, risk assessment/analytical design space, and analytical control strategy.
Method performance requirements (or design intent) are based on a fundamental understanding of the criteria the method must meet, the CQAs, and acceptance criteria. Method development (design selection) is a critical decision and requires understanding of the CQAs and the intended use of the method.
Defining the design space accurately ensures that the method or methods will generate accurate, reliable data and meet all performance criteria under all anticipated conditions of use as the method winds its way through its lifecycle.
Risk assessment employed during design space evaluation helps to identify where variability may jeopardize the ability of a method to deliver as intended. Risk assessment includes robustness studies for highest-risk parameters, including design of experiment for assessing multidimensional combinations and interactions of the highest-risk factors.
Previously identified critical parameters and associated acceptable ranges must be defined during method control (also called control verification). One must also characterize the method system suitability, which encompasses acceptance criteria ensuring that the method is performing as it should. Some groups also perform instrument checks, but these are often unnecessary since method suitability should already have provided that information previously. Finally methods are evaluated to confirm their suitability for intended use. This may take the form of a formal qualification, validation, or verification.
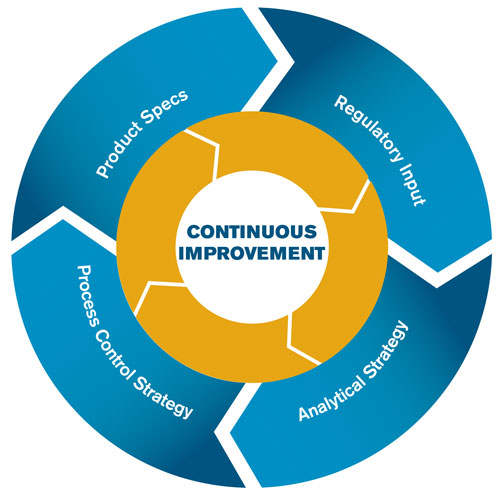
Process and analytical control, product specifications, and regulatory input are intricately linked and harmony between all of them is essential to continuous improvement.
Method Lifecycle Management
Applying QbD to analytical method development and establishing a control strategy are merely starting points for the real challenge: managing and updating methods as the process evolves.
The first step is to monitor method performance continuously through control samples. The methods themselves also require periodic evaluation. Under a traditional paradigm the method does not change, nor are potential adjustments considered, between qualification and validation. Analytical groups should instead take advantage of additional information that becomes available, and use it to determine if the method truly is performing as intended.
The thoroughness of this exercise should be commensurate with the criticality of the development/production phase. Light validation or qualification will usually serve during Phase I studies, while Phase II demands more involved assessment of previously generated data. Phase III requires full validation per USP or ICH guidelines.
Similarly, method robustness requires continuous assessment during method development (not just during the analytical team’s learning curve). Assessment should include performance when parameters—for example column temperature or flow rate in an HPLC analysis—are deliberately adjusted or changed.
Robustness may be assessed during method validation as well, though much simpler is to examine it at strategic junctures during development. Since validation is essentially a confirmation that the method works, it is an inappropriate point at which to screen method conditions or gain additional performance knowledge. Evaluating ruggedness should similarly be continuous.
Analytical methods and product specifications are intimately connected, and this needs to be understood as specifications are established or revised. A QbD approach helps to align thinking between process and analytical groups, and facilitates their continuous communication.
Andre Johnson ([email protected]) is associate director of biotech services at Covance. Web: www.covance.com.

