
October 1, 2012 (Vol. 32, No. 17)
Recent strides in biotechnology have provided a wealth of information about the genomes of many species, some of which were challenging or impossible to interrogate with approaches that were dominating the field merely a few years ago.
As a result, unprecedented opportunities became available, including the possibility to perform genome-wide gene expression profiling and comparative genomic analyses, and to study changes that occur in specific states, such as during development, disease, or in response to environmental agents or therapeutic compounds.
Historically, many biological questions were addressed by relying on datasets that originated from large-scale gene expression studies. However, one of the shortcomings of this strategy is that gene expression heterogeneity, which is present even among genetically identical cells that are grown in the same culture or are found at the same developmental point, is obscured, and information about individual cells and cell-to-cell variability will only be captured by performing single-cell level analyses.
“The very next frontier for genomics, as a field, is single-cell genomics, and the expression profiling of single cells will likely be the next step,” says Jian-Bing Fan, Ph.D., senior director of scientific research at Illumina.
To gain information about transcriptional profiles at the single-cell level, investigators in Dr. Fan’s lab, in collaboration with researchers from the Karolinska Institute led by Sten Linnarsson, described a highly multiplexed genome-wide strategy using single-cell RNA-Seq.
In this protocol, ideally suited for single-cell analysis, 96 cells placed into the individual wells of a 96-well plate were simultaneously lysed. The mRNAs were converted to cDNAs, and after barcodes and primer binding sequences were introduced by the template-switching activity of reverse transcriptase, the cDNAs were pooled and processed for 5´-end sequencing on the Illumina next-gen sequencing platform. This technique can use as little as a few picograms of starting mRNA, and is appropriate for the transcriptional profiling of rare cells.
Dr. Fan, in a collaboration with Kun Zhang, Ph.D., at the University of California, San Diego, is currently developing a strategy to generate a high-resolution three-dimensional map of transcriptional activities in single cells. In the past, after being isolated, human tissue samples were trypsinized and the resulting cell suspensions were used to examine individual cells. But during this procedure, the spatial relationship among cells from the original tissue sample was lost.
“An important aspect of the technology that we are working on is the additional layer of information that it provides. It does not require prior knowledge about the identity of the specific cell types and will be fundamental for understanding cellular functions that shape development and disease,” says Dr. Fan.
While data analysis promises to be challenging, and might require the development of new types of protocols, a new level of spatial organization that has not been available before is about to emerge.
“Mapping the specific individual cells back to the three-dimensional organ or tissue is challenging, but this is the future direction into which single-cell gene expression profiling will develop,” explains Dr. Zhang.

High-throughput screens help researchers better understand the regulation of gene expression as well as the role of gene deregulation in causing disease. [Luchschen/ShutterStock]
High-Resolution 3D Map
Recently, particularly with the developments in the field of epigenetics, the interface between DNA methylation and gene expression has become a vibrant research topic. For many years, it has been known that genes can become switched off by abnormal methylation in cancer. Subsequently, studies that examined the methylome identified many genes that exhibit aberrant methylation in cancer cells, when compared to their untransformed counterparts.
“Most of these events are probably irrelevant and, in all likelihood, they represent the consequence of the cell being a cancer cell, just as it also happens with mutations, many of which are known to be passengers,” says Peter A. Jones, Ph.D., D.Sc., distinguished professor of urology and biochemistry at the University of Southern California.
While the presence of aberrant DNA methylation changes in cancer cells has become relatively easy to explore, a much more challenging aspect is to determine whether these methylation changes are driving the process of malignant transformation or whether their emergence is consequential to it. Dr. Jones and colleagues recently described an approach that facilitates the identification of the epigenetic driver events required for cancer survival.
The investigators hypothesized that the few key chromosomal regions whose methylation is required for cancer cell survival would preferentially remain methylated when global methylation levels are reduced. After surveying residual DNA methylation levels in cancer cells with genetically disrupted DNA methyltransferases and clustering profiles from normal cells and primary cancer tissues, Dr. Jones and colleagues used gene expression meta-analysis to define the regions that are dependent on DNA methylation-dependent gene silencing.
Their strategy helped identify a set of promoters whose methylation is required for somatic cancer cell survival. Some of the respective genes are shared among several tumor types, while others are cancer- or tissue- specific. “The genes identified by this approach have not been previously known as tumor suppressor genes,” notes Dr. Jones.
In addition to helping define and validate the minimal DNA methylation profile that is necessary and functionally relevant for cancer cell survival, this approach advances our knowledge on the molecular events that drive cancer development, and promises to define a new generation of epigenetic therapies, in which the alterations that are being targeted are exactly the ones that the cancer cells rely on for survival.
While over the past 15 years the field of DNA methylation has focused on promoters, there are additional regions where methylation, even though not well understood functionally, could play important biological roles.
“We are looking more globally to see whether the bodies of the genes and the enhancers, which also undergo methylation, but we do not know yet it role at these locations, is important. It is important to look beyond promoters,” says Dr. Jones.
A significant challenge in biomedical sciences emerged when functional genomics developments, which uncovered novel aspects about biological systems, have not been paralleled in pace by drug discovery advances.
“Another problem is that the current strategy of optimizing candidate therapeutic compounds, which relies on a single target, is somewhat dangerous, and measuring the collective response of all relevant genes related to a specific phenotype, which is a much better option, has been technologically challenging,” says Xiang-Dong Fu, Ph.D., professor of cellular and molecular medicine at the University of California, San Diego.
RASL-Seq
To address this challenge, Dr. Fu and colleagues recently developed and described RASL-Seq, a new massively parallel quantitative gene analysis strategy in which RNA-mediated oligonucleotide annealing, selection, and ligation are coupled with next-generation sequencing, allowing hundreds of genes to be followed concomitantly under thousands or tens of thousands of distinct conditions.
RASL-Seq helped investigators in Dr. Fu’s lab develop a high-throughput sequencing method, which allows multi-target drug design to be performed in a pathway-centric, high-throughput fashion, a strategy neatly designed for the study of small molecules that affect disease-specific gene expression pathways. The fact that specific drug targets do not need to be identified in advance represents one of its advantages over conventional chemical screening.
Dr. Fu and colleagues illustrated the power of this technique in a study that sought to identify small inhibitors of androgen receptor-mediated gene expression for treating hormone-refractory prostate cancer and identified several classes of compounds. One of them, a cardiac glycoside, inhibited both androgen-sensitive and androgen-resistant prostate cancers without exhibiting severe cytotoxicity, and this effect most likely occurred through the proteasome-dependent degradation and destabilization of the androgen receptor.
“This approach brings a unique advantage when studying complex diseases because of the multitude of pathways that are involved. It may contribute to an integrated pattern of gene expression,” explains Dr. Fu.
In another study using RASL-Seq, investigators in Dr. Fu’s lab blocked different branches of EGF-induced signaling and unveiled a serine/arginine protein-specific kinase (SRPK)-dependent signal transduction pathway with a central role in regulating alternative splicing in mammalian cells.
“This is an intriguing research area, because many diseases are known to be caused by defects in splicing regulators, but very few signaling molecules involved in regulated splicing have been characterized to date,” says Dr. Fu.
“In many diseases, even though we can identify the environmental input linked to pathogenesis, we do not know how it contributes to the pathological modifications,” notes Feng C. Zhou, Ph.D., professor of anatomy, cell biology and neurobiology at the Indiana University School of Medicine.
Fetal Alcohol Disorder
An interesting example is provided by the fetal alcohol spectrum disorder, a multisystem condition that is causally linked to maternal alcohol use during pregnancy, and is characterized by physical, behavioral, and cognitive deficits, including developmental delay, growth deficiency, craniofacial dysmorphology, and modifications that affect other organs and systems at various degrees.
Previously, Dr. Zhou and colleagues revealed that ethanol exposure alters the cellular DNA methylation program during early neural tube development, and identified over 2,100 epigenetically changed genes in which the cytosines are differentially methylated in alcohol-treated embryos.
To better understand the protein expression changes that occur as a result of epigenetic and genetic changes in the fetal alcohol spectrum disorder, Dr. Zhou, in collaboration with Stephen Mason, Ph.D., an adjunct researcher who is also in the department of anatomy and cell biology at the Indiana University School of Medicine, used whole-embryo cultures to examine the alcohol-signature protein profile across all cell and tissue types in mice at the early neural developmental stage during neurulation.
“This study is a continuation of our previous studies,” explains Dr. Zhou.
The team identified 40 protein spots that were differentially expressed between alcohol-treated and control cultures. Several of these proteins, confirmed by mass spectrometry, fulfill key roles in the cell cycle and the ubiquitin-proteasome pathway.
The results indicated that epigenetic and genetic changes occurring as a result of alcohol exposure impact protein expression during neurulation. The biological functions that were perturbed were linked to tissues and organs that originate, during development, from all three embryonic layers.
“Analyses of epigenetic modifications and those that survey gene expression, protein expression, and metabolic perturbations are required to investigate the molecular and cellular changes as a whole, to eventually understand the causal mechanisms that differ between two distinct states, such as between the child with developmental delay caused by drinking during pregnancy and the healthy child,” explains Dr. Zhou.
“We are trying to understand how gene expression is regulated, how its deregulation causes disease, and ultimately how we can correct those deregulated states to treat disease,” says Bing Zhang, Ph.D. assistant professor of biomedical informatics at the Vanderbilt University School of Medicine.
Dr. Zhang’s group has explored the possibility of defining a gene expression signature that can be used to make prognostic and therapeutic decisions in patients with colorectal cancer. This malignancy, currently the third leading cause of cancer mortality worldwide, is stratified into four stages (I to IV), with a higher stage being assigned to more serious disease.
Cure, which occurs in approximately 95% of patients with stage I disease treated surgically, is difficult to accomplish in patients with stage IV disease, who also require chemotherapy.
“For stage II and III patients, which represent a large population, the question is whether we need to provide chemotherapy, because previous clinical trials suggest that it is not required for certain patients, but it is beneficial for others,” explains Dr. Zhang.
Histological features, such as tumor size, lymph node positivity, and metastatic dissemination have been used to perform such predictions in the past, but often they were not reliable.
“Our idea was to use gene expression analysis to better predict the prognosis of stage II and III colon cancer patients,” explains Dr. Zhang.
A challenging aspect is that different gene expression signatures from different studies often do not overlap with each other. “We tried to find common biological themes across previously published signatures, and by integrating them into biological networks, we generated a gene expression signature that is more biologically meaningful and, additionally, has a good prognostic value,” says Dr. Zhang.
This approach revealed that genes with mechanistically important roles in colorectal cancer may be used to develop reliable prognostic models that accurately predict recurrences and the response to adjuvant chemotherapy. Gene expression-based stratification of these two cancer stages is crucial for increasing survival and the quality of life, while minimizing the chemotherapy-associated toxic effects and the financial costs involved.
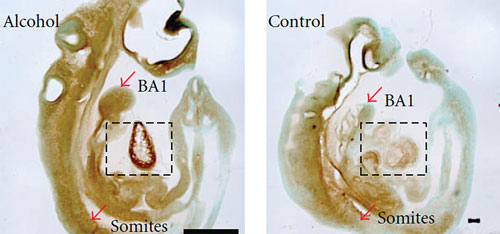
Research has shown that alcohol induces aberrant changes in gene expression and in the epigenome of embryos. One result is the misregulation of proteins such as serotransferrin, which was found in greater amounts in mouse embryos exposed to alcohol. The increase (A) was confirmed by immunolocalization of serotransferrin to the branchial arches (BA1) and somites, particularly in the heart (boxed area), as compared to control (B). Misregulated proteins are known to be involved in nervous system development and function. [International Journal of Proteomics, Hindawi Publishing, 2012, doi:10.1155/2012/867141]
3D Systems
While historically the biology of specific cell types was examined by using homogenous monolayer cultures grown in vitro, this experimental approach tends to be an oversimplification. As opposed to the culture systems that often use a single cell type, organs consist of a complex network of multiple cell types that also includes extracellular matrix, growth and signaling factors, and other components.
Dynamic bidirectional signaling events within this microenvironment assume key functions in tissue maintenance and in suppressing or promoting tumor development. The microenvironment is one of the most important factors that emerged in recent years to shape cellular behavior, and three-dimensional (3D) cell culture systems, which more faithfully reflect the in vivo conditions, have increasingly found applications in many biomedical areas.
Nils Cordes, M.D., Ph.D., professor of molecular and cellular radiobiology at the Dresden University of Technology, and colleagues have comparatively examined the gene expression profiles in two exponentially growing human cancer cell lines—lung carcinoma and squamous cell carcinoma—grown in a 3D extracellular matrix scaffold or as a conventional two-dimensional monolayer.
“The idea behind this experiment was that in vitro cell cultures that we are working with do not always reflect the in vivo situations, and we wanted to know whether the differential gene expression shapes tumor cell sensitivity to radiation and chemotherapeutics,” explains Dr. Cordes.
The technique revealed significant gene expression changes between the two conditions.
“We showed that there was a differential gene expression pattern, between the 3D and the 2D conditions, in genes encoding ECM proteins, integrins, cell shape proteins, and proteins linked to morphology, but not to DNA repair,” notes Dr. Cordes.
Moreover, under the 3D growth conditions, cells showed an increased resistance to radiotherapy and chemotherapy as compared to the 2D conditions, pointing toward the importance of the growth conditions in shaping gene expression. Important in this context is another study from Dr. Cordes’ group, in which differences in cell cycling, oxygen levels, and radiation dosimetry were ruled out as critical determinants of higher tumor cell survival levels under 3D growth conditions.
Few advances have reshaped biomedical and clinical areas within such a short time, and to such a great extent, as the ones that marked gene expression analysis. Technological developments, their intimate integration with many basic science disciplines, and a growing number of clinical applications have been defining this field for the past decade.
While collecting increasingly sophisticated and complex datasets has undoubtedly played an instrumental role, perhaps an even greater impact was exerted by the development and implementation of novel conceptual frameworks, a theme that provides a central learning point and was so relevantly expressed in Sir William Bragg’s words: “The important thing in science is not so much to obtain new facts as to discover new ways of thinking about them.”

