April 1, 2015 (Vol. 35, No. 7)
To Detect Oncogenic Shifts in Expression, It Helps to Survey Subtle RNA-Level Processes
Analyzing the entire cancer transcriptome or targeted transcripts with deep genetic profiling and other technologies provides a rapid and accurate means to detect potentially important expression changes. Further, identifying and evaluating modifications in the RNA of tumors helps reveal not only their oncogenic properties, but also how they will respond to anticancer therapies.
Nucleotide modification is one of the most evolutionarily conserved properties of RNA, according to Christopher E. Mason, Ph.D., an assistant professor of computational biomedicine at Weill Cornell Medical College.
“There are many ways disruption of RNA can contribute to cancers,” says Dr. Mason. “This can occur immediately after being synthesized, localization, or later from defects in processing.”
An example of RNA acting as an immediate oncogenic force, continues Dr. Mason, may be seen in some of the thyroid cancers found in children who were exposed to fallout after the Chernobyl nuclear reactor accident: “In our collaborative study, 22 tumors were found to harbor fusion oncogenes, many of which aberrantly activated MAPK signaling pathways. Additionally, other fusions led to overexpression (that is, enhanced transcription) of a nuclear receptor associated with malignancy.”
Issues also can arise from RNA processing defects. “Examples include retention of introns or failures in spliceosome functions,” notes Dr. Mason. “In the cytoplasm, RNA editing process may also become dysfunctional. Perhaps an A is inserted instead of a G, and what was benign now becomes oncogenic.”
Researchers are exploring a range of strategies that focus on tapping into the power of the human immune system to battle a range of diseases, particularly cancer. [Freshidea/Fotolia]
Epitranscriptome
A new and exciting area of research is what Dr. Mason calls the “epitranscriptome,” which describes the multitude of RNA modifications. “There are currently about 110 known RNA base modifications,” Dr. Mason points out. “Alterations in methylation can pathologically change where RNA goes, how its information is interpreted, or create changes in its stability. All of these can contribute to the development of cancer.”
Coupling information from the epitranscriptome with a wealth of other contemporary technologies is allowing new ways of investigating cancer. In a recent study of acute myelogeneous leukemia (AML) and chemoresistance, Dr. Mason and colleagues found a convergence of disrupted biological networks in DNA, in DNA methylation, and in RNA. They identified new prognostic indicators that appeared to be associated with predicting relapse time, and they observed allele-specific switching of damaging mutations. Their findings suggest a dynamic evolutionary landscape of complex molecular changes that promote tumor survival.
Dr. Mason says it is an exciting and extraordinary time in scientific research: “We are just beginning to understand the transcriptomes of cancer. This will allow us to better discover and design drugs more quickly and that work with greater efficacy.”
RNA Editing and Cancer
A fundamental biological concept is that transcribed RNA has the exact same corresponding sequence as its original DNA template. New studies show this is not quite the way Mother Nature operates. Adenosine deaminases acting on RNAs (ADARs) can edit nucleotides by changing an adenosine (A) into an inosine (I) that is subsequently read by cellular machinery as guanosine (G).
“RNA editing is an epigenetic control mechanism that until recently was thought to be a rare occurrence,” reports Jin Billy Li, an assistant professor of genetics at the Stanford University School of Medicine. “It is becoming increasingly clear that there actually are many RNA editing sites, and that many of these are very likely functional. A-to-I editing may occur at more than one hundred million genomic sites that are located in a majority of human genes.
“RNA editing is an evolutionarily efficient approach to make changes in proteins by providing isoforms with different functions. Further, tissues from the same person often differ in their RNA editing profile.”
According to Dr. Li, recent studies suggest that editing enzymes may be more highly expressed in cancer tissues. He adds that the introduction of rapid means of genome and transcriptome sequencing has made it possible to begin to look at how RNA editing may contribute to cancers.
“A study [by Chen et al. that appeared in Nature Medicine in 2013] utilized transcriptome sequencing and found that A-to-I editing of AZIN1 (encoding antizyme inhibitor 1) was elevated in human hepatocellular carcinoma,” details Dr. Li. “The editing caused a serine to glycine substitution that resulted in mislocalization and a gain-of-function of the protein that enhanced its tumor initiating potential.”
Since the pattern of RNA editing may differ in the tumor versus its neighboring normal tissues, it is important to sequence both to identify malignant-inducing changes. The new field of RNA editing adds a layer of complexity in identifying cancer-causing mutations.
“Projects such as The Cancer Genome Atlas (TCGA), which provides for large-scale sequencing of cancers and dissemination of data to investigators, will help accelerate our knowledge of such changes,” asserts Dr. Li. “In the future, better understanding of RNA-editing mechanisms will likely help revolutionize how we evaluate and treat different types of cancer.”
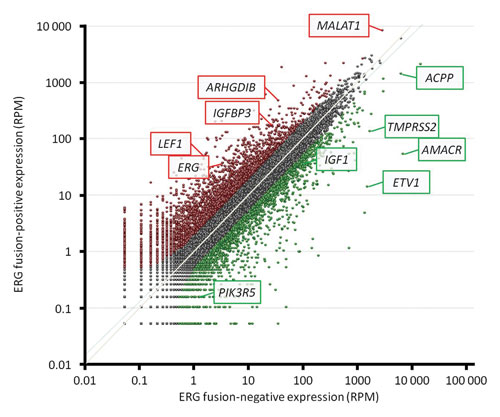
Scatterplot of expression of transcripts identified by whole-transcriptome sequencing. The 5,885 genes that were differentially expressed between the two samples are marked with red dots, if more highly expressed, and green dots, if less highly expressed, in the fusion-positive sample. Genes of particular interest are highlighted. RPM: Reads per million; ACPP: acid phosphatase, prostate; AMACR: a-methylacyl-CoA racemase; ARHGDIB: Rho GDP dissociation inhibitor ß; ERG: ets-related gene; ETV1: ets variant 1; IGFBP3: insulin-like growth factor-binding protein 3; IGF1: insulin-like growth factor 1; LEF1: lymphoid enhancer-binding factor 1; MALAT1: metastasis-associated lung adenocarcinoma transcript 1; PIK3R5: phosphoinositide-3-kinase regulatory subunit 5; TMPRSS2: transmembrane protease, serine 2. [Anticancer Research, Vol. 32, No. 9, September 2012]
Mutation Signatures
The editing of DNA also drives genetic diversity. Families of evolutionarily conserved enzymes assist in this process by catalyzing DNA cytosine-to-uracil deamination. The APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like) family of DNA-editing enzymes is made up of nine active members. These enzymes contribute broadly to innate antiviral immunity, but specific members also function to diversify antibody gene DNA (AID) as well as edit mRNA transcripts (APOBEC1).
A well-studied example is the APOBEC targeting of retroviruses such as HIV-1. During reverse transcription, HIV cDNA intermediates are vulnerable to enzymatic deamination. At least four different APOBEC proteins are capable of attacking HIV-1 cDNA during this susceptible phase of the viral life cycle. But it takes only one member to spoil the family’s reputation. Such is the case for APOBEC3B. This enzyme has been implicated as a source of mutations in breast, lung, cervical, head and neck, ovarian, and bladder cancers.
Reuben Harris, Ph.D., a member of the Masonic Cancer Center and a professor of biochemistry, molecular biology, and biophysics at the University of Minnesota, is an expert on both immunity and cancer functions of the APOBEC family. In 2013, in the journal Nature Genetics, his laboratory reported that APOBEC3B is upregulated in breast tumors and correlated with higher levels of both cytosine-to-thymine and overall base-substitution loads.
Dr. Harris hypothesized that APOBEC3B might be a common mutagenic factor widely affecting genesis and evolution of many different types of cancers. To address this idea, his laboratory performed a comprehensive unbiased analysis of all available DNA deaminase expression profiles and cytosine mutation patterns in 19 different types of cancers. They analyzed expression data, base-pair mutation frequencies, local cytosine mutation signatures, overall mutation loads, and localized areas of hypermutation. Taken together, these studies strongly implicated APOBEC3B in the mutagenesis of multiple cancer types.
“Independent work from other [research teams] including [those led by] Drs. Dmitry Gordenin and Mark Stratton arrived at the same conclusion,” stated Dr. Harris. “Therefore, it is now clear that APOBEC3B contributes to ongoing mutagenesis in many different cancer types. This information may be used in the future to identify mutator cancers more liable to therapeutic failure and that these will require special tailored treatments.”
Colon Cancer
MicroRNAs are small endogenous activators of the RNA interference (RNAi) pathway. These endogenous products of noncoding genes (~22 nucleotides) function to degrade, destabilize, or sequester target mRNAs, thus inhibiting translation. Appropriate function of microRNAs is crucial for normal tissue homeostasis.
Many, though, are located in fragile regions of the genome, and their expression is altered with the progression of cancers. Amanda B. Hummon, Ph.D., an assistant professor of chemistry and biochemistry at the University of Notre Dame, is studying expression of specific microRNAs in colon cancer.
“Colon cancer is the third leading cause of cancer death. It is estimated that 100,000 new cases and 50,000 deaths occur each year. Among others, expression of two microRNAs, miR-143 and miR-145 is significantly reduced in colon cancer,” says Dr. Hummon. “These two nonhomologous microRNAs cluster in the 5q32 chromosome and directly target several specific mRNAs. We wanted not only to know whether it is better to study clustered microRNAs as a group or individually for the greatest information.”
Dr. Hummon and colleagues profiled the expression levels using qRT-PCR of both microRNAs in five human colon cancer cells lines and six pairs of patient-matched FFPE colon tissue samples. These results validated their lowered expression.
The investigators followed up by combining SILAC assessment and microarray analyses to systematically study the impact of miR-143/-145 on the colon cancer proteome and transcriptome. SILAC, stable isotope labeling with amino acids in culture, is a mass spectrometry technique that provides a means to track protein abundance using nonradioactive protein labeling.
“Our results gave us access to the cancer transcriptome and indicated that it is necessary to investigate both the individual and combined functional implications of a microRNA cluster. Examining individually introduced microRNAs gave a pattern distinct from that of the assembled cluster,” explains Dr. Hummon. “This finding has broad implications to additional clusters in a number of disease states and cellular processes. We now want to see if these findings can be extended to other microRNA clusters and are expanding our studies to examine the miR-23a~27a~24-2 cluster.”
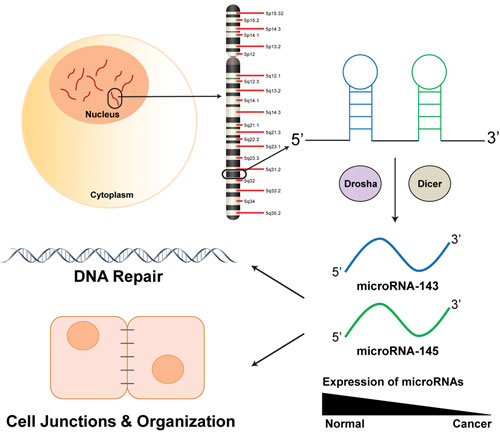
MicroRNA-143 and microRNA-145 exist in a cluster on chromosome 5 in humans. Their expression is decreased in colorectal cancer tumors, resulting in aberrant functioning of many cellular path-ways. Two critical cellular functions of microRNA-143 and microRNA-145 were found by researchers at the University of Notre Dame to play a role in DNA repair pathways as well as in cellular junctions and organization.
SKIP and RNA Splicing
The multiprotein enzyme complex called RNA polymerase II catalyzes the transcription of DNA. However, it often stalls in the process and requires the help of elongation/splicing factors such as SKIP, the Ski-interacting protein. Katherine A. Jones, Ph.D., a professor in the Regulatory Biology Laboratory of the Salk Institute for Biological Studies, found that SKIP is also involved in survival of cancer cells. Dr. Jones’ group is interested in better understanding the intricate links between transcription elongation and RNA processing and how these are involved in cancers.
“We utilized genomic and proteomic approaches to understand this process more clearly. Our studies employed RNAi chromatin immunoprecipitation and RNA immunoprecipitation to show that SKIP recruits a critical splice factor to the p21Cip1 gene, indicating it is critical for both splicing and expression of this cell cycle arrest factor,” asserts Dr. Jones. “These studies also demonstrated that SKIP is needed for both stress-induced and basal expression of p21Cip1.”
Dr. Jones next wanted to know if loss of SKIP would sensitize cells to apoptosis induced by chemotherapeutic agents that are known to damage DNA. Her group, she said, “found that depleting SKIP induced a rapid down-regulation of p21Cip1 and also predisposed cells to undergo p53-mediated apoptosis, which was most pronounced in cells also subjected to DNA damage.”
These findings, says Dr. Jones, open the door to seek small molecule inhibitors of SKIP: “Since chemotherapeutic agents that damage DNA can have increased potency when SKIP is depleted, identifying inhibitors of SKIP might boost the efficacy of killing cancer cells by chemotherapeutic agents.”
Technological advances continue to make headway in the speed and accuracy of profiling the cancer transcriptome. This is allowing unprecedented levels of analyses of individuals and groups with specific cancers. These rapidly evolving investigations are also setting the stage for new biomarker discovery and more personalized treatments.

