
January 15, 2013 (Vol. 33, No. 2)
The fascination with epigenetics stems not only from the profound impact that it has exerted on the biomedical, medical, and social sciences, but also from the somewhat debated and elusive definition of the term itself. It’s shifted multiple times over the years.
A key feature of epigenetic changes— their potential heritability—brings new dimensions to an already vibrant and thought-provoking field, but this aspect has received relatively little attention until recently. One of the prerequisites for epigenetic inheritance is that a specific gene expression pattern be re-established after DNA replication, in daughter cells, to ensure the faithful inheritance of the chromatin architecture.
“Until now, all our knowledge about the inheritance of epigenetic markings has been largely hypothetical, because no methods were available to look at what is happening with proteins just after DNA replication,” says Alexander M. Mazo, Ph.D., professor of biochemistry and molecular biology at the Jefferson Medical College, Thomas Jefferson University.
Previously, it was proposed that histone post-translational modifications were the ones responsible for epigenetic inheritance, but to a large extent these models were based on theoretical assumptions and were not fully supported experimentally. As an additional shortcoming, they did not clearly explain the ability of histones, which cover huge regions of DNA, to recruit their binding partners in a sequence-specific way.
By surveying in vivo protein-protein interactions on nascent DNA sequences at replication forks and examining their posttranslational modifications, Dr. Mazo and colleagues showed that trimethylated H3K4 and H3K27 are replaced by unmethylated histones after DNA replication and are not transferred from parental to daughter nucleosomes.
“It seems that the current model, that methylated histones are stably associated with DNA during replication and are transferred from parental to daughter strands, may not be true, but certain histone-modifying proteins seem to be working as epigenetic marks,” continues Dr. Mazo.
Enhancer-of-Zeste, an H3K27 methylase, Trithorax, an H3H4 methylase, and Polycomb were stably bound to nascent DNA, and their association with the nascent DNA was constitutive and continuous through the S phase, in the absence of trimethylated histones. “Our findings also revealed that epigenetic markings are present at very specific regions, which is one of the key conditions for epigenetic inheritance,” explains Dr. Mazo.
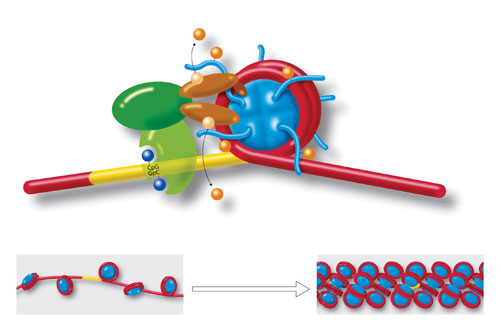
Molecules and a DNA strand loop around a cylindrical histone core (blue), to form a nucleosome. Yellow region is a section of cytosine-guanine (CpG-GpC) di-nucleotides that play a role in chromatin formation. Also shown are acetylation, deacetylation, methylation, and demethylation. Strings of nucleosomes (lower left) form the structure called “beads on a string.” Further compacting (lower right) condenses the chromatin even more to fit inside cell nuclei. Chromatin organization and dynamics, shaped by developmental and environmental influences, have attracted much attention as crucial facets of epigenetics. [Art for Science/Photo Researchers]
Epigenome-Wide Association Studies
Since the first genome-wide association study in 2005, over 1,400 articles were published, reporting almost 8,000 SNPs.
Although genome-wide association studies revolutionized our understanding of the genetic basis of complex diseases and traits, the relatively small effect exerted by each allele, and the small proportion of the heritability that they explain collectively, caused some disappointment.
“That has created discussions on missing inheritance and possible nongenetic contributions, where 80–90% of the disease phenotype cannot be explained by all the genetic variants that have been identified so far,” says Stephan Beck, Ph.D., professor of medical genomics at the University College London Cancer Institute.
Complementing these observations, studies on monozygotic but disease-discordant twins also revealed that on average only about 50% of the phenotype can be explained by genetic changes.
“These are essentially the reasons why we have set out the concept of epigenome-wide association studies, to track down the missing factors that must contribute to these diseases but cannot be explained by genetic variations,” explains Dr. Beck.
The principle of epigenome-wide association studies involves scanning cases and controls to identify epigenetic variations associated with a specific trait or disease. Although similar to genome-wide association studies, two major differences need to be considered.
One is that unlike in genetic studies, where peripheral blood DNA can serve as a surrogate for all tissues, the highly tissue-specific nature of epigenetic modifications cannot be captured by using a single source of DNA.
“One has to chose the source of the study material very carefully, to make it informative for the phenotype to be studied,” explains Dr. Beck.
The second aspect is that, unlike in genetic studies, where interpreting a mutation is based on the knowledge that common diseases are not genotoxic and do not change the gene sequence, an association found in epigenetic studies is not necessarily the cause of the phenotype, but may be the consequence of it, a phenomenon known as reverse causation.
Therefore, it is not suitable to simply analyze a single case/control cohort, as in genome-wide association studies.
“We suggested a powerful two-tiered study design, in which one should first look at disease-discordant monozygotic twins to exclude genetic changes, and after identifying epigenetic differences, verify and replicate them in unrelated individuals that have been prospectively followed,” explains Dr. Beck.
Integrate Studies with Approaches
The prospective study design ensures that samples are collected and examined before and after a phenotype develops, allowing reverse causation to be excluded.
“An important point, now that we have an additional way to analyze common disease variation, is that we should not replace, but integrate epigenetic studies with genetic approaches, and together they should provide more explanations of what could cause the disease,” emphasizes Dr. Beck.
“Over the last couple of years, a revolution in our ability to not only sequence, but also synthesize vast amounts of DNA, has enabled us to study the relationship between DNA sequences, epigenetic marks, and gene regulatory activities in a directed and hypothesis-driven manner,” notes Tarjei S. Mikkelsen, Ph.D., principal investigator at the Broad Institute and Harvard Stem Cell Institute.
Dr. Mikkelsen and colleagues used a strategy that combines bioinformatics, synthetic biology, and experimental approaches to examine histone methylation changes that occur over time during the differentiation of human mesenchymal stem cells into adipocytes.
The genome-wide chromatin state maps that were created allowed the dynamic chromatin signatures characteristic for specific stages during differentiation to be visualized and facilitated the identification of key regulatory elements.
“This strategy is very informative for identifying active gene promoters and other functional elements in the genome in a context-dependent way,” explains Dr. Mikkelsen.
In a subsequent study, Dr. Mikkelsen and colleagues designed a massively parallel reporter assay to facilitate the functional analysis of individual regulatory sequences from the human genome at a higher resolution than currently existing approaches. This strategy, which can be adapted to other experimental settings, involves the synthesis of tens of thousands of tagged oligonucleotides that contain a library of regulatory elements.
Each oligonucleotide is cloned on a plasmid containing an optional promoter, a regulatory element, and an open reading frame. After transfecting the plasmid pool into cells, the tags on the reporter mRNAs are sequenced and counted to determine their relative activities.
“We can generate many carefully defined mutations of a natural enhancer and determine in parallel, using the sequencing readout, how each mutation changes its activity,” continues Dr. Mikkelsen.
Investigators in Dr. Mikkelsen’s lab illustrated the strength of this strategy with two inducible enhancers, a synthetic cAMP-regulated enhancer, and a virus-inducible enhancer of the human interferon beta gene. After mapping the transcription factor binding sites at single-nucleotide resolution, quantitative models helped identify mutations that increase enhancer inducibility.
Going forward, they plan to insert these synthetic sequences into the genome to examine how they interact with nearby epigenetic marks.
“We still do not understand whether the genetic information always determines the epigenetic landscape, or whether there is inheritance at the epigenetic level that has no basis in the genetic information. The synthetic biology approach is emerging as a very powerful tool for probing these questions,” says Dr. Mikkelsen.
Blueprint Project
“DNA methylation is like a storyteller. We believe that it keeps a memory of the cell of origin during development and it is informative about activities that take place during tumor development,” says Jose Ignacio Martin-Subero, Ph.D., principal investigator and leader of the epigenomics group at the University of Barcelona.
As part of a collaborative endeavor established by two major initiatives, the EU-funded Blueprint Project and the chronic lymphocytic leukemia (CLL) genome project, Dr. Martin-Subero and colleagues examined DNA methylation changes in 139 patients with CLL. The availability of the methylome, exon sequencing, and gene expression profiling data for this patient group, and access to their clinical reports, provided an unprecedented opportunity to examine methylation, gene expression, and clinical parameters in parallel.
One of the features that made this study unique is that, instead of using normal B cells as controls, it compared the full methylomes between pure populations of naïve and memory B cells, two subtypes that were isolated from the peripheral blood of a single donor.
“We found approximately 1.7 million CpG sequences that were significantly and clearly differentially methylated, indicating that a massive modulation of the DNA methylome occurs during B-cell differentiation, a finding that was quite unexpected,” notes Dr. Martin-Subero.
Patients with CLL belonging to one of two subtypes, either with very favorable prognosis, related to memory B-cell origin, or with a slightly worse prognosis, related to naïve B-cell origin, exhibited very distinct DNA methylome signatures, despite expressing very similar gene sets, and DNA methylation did not correlate well with gene expression.
A comparison revealed that approximately 90% of the changes were hypomethylation, which was enriched in gene bodies and intergenic regions, while the less frequently occurring hypermethylation was enriched at transcriptional start sites.
By using the ENCODE data to examine the genomic distribution of hypomethylation, the investigators found an enrichment of this modification at enhancer regions during both B-cell differentiation and CLL pathogenesis. In addition, a consensus cluster analysis that used 10,000 permutations of the slightly over 1,600 CpG sites found that approximately 15% of the patients did not belong to any of the two groups, but showed an intermediate DNA methylation profile, and had an intermediate prognosis.
“We believe that DNA methylation helps us better classify this disease, and the cell of origin seems to be associated with prognosis, because the more undifferentiated it is, the worse the prognosis seems to be,” explains Dr. Martin-Subero.
While alternative splicing was estimated to occur in approximately 95% of human genes, and has increasingly been implicated in gene regulation during development and disease, understanding the propagation and maintenance of alternative splicing patterns during cell division and differentiation remains elusive.
“Alternative splicing is the last of the major steps in gene expression that we still do not understand, and the idea that these patterns could be determined by epigenetic marks is very attractive,” says Tom Misteli, Ph.D., head of the ccll biology and genomes group at the National Cancer Institute.
Recent Model
A model that emerged several years ago examined splicing patterns for the same alternatively spliced reporter gene that was expressed from promoters of variable strength, and proposed that splicing is shaped by the RNA polymerase elongation rate. More recently, several groups found, during genome-wide histone modification analyses, that specific histone modifications accumulate over exons, marking their boundaries.
“This was intriguing as it drew a lot of attention to the idea that chromatin and epigenetic marks could affect splicing and brought the two areas together,” says Dr. Misteli.
Recent work in his lab made important contributions toward unveiling the interface between splicing and epigenetic modifications. In a survey of histone modifications on the alternatively spliced FGFR2 gene, Reini Luco, Ph.D., at the time a post-doctoral fellow in the Misteli lab, found that enrichment in a particular histone modification, H3K36 trimethylation, recruits, through a protein adaptor protein, a polypyrimidine tract binding protein that suppresses the inclusion of an exon, leading to mesenchymal stem cell splicing patterns.
The alternative outcome, in which recruitment of another chromatin factor causes the exclusion of a different exon, specifies epithelial stem cell lineage commitment.
“Additional layers of regulation most likely exist, because we know, for example, that if certain splicing factors are phosphorylated, their propensity for being recruited to RNA molecules changes,” explains Dr. Misteli.
As a field that unveils new concepts and complements genetic and genomic approaches, epigenetics promises a novel framework to dissect cellular and molecular networks shaping development, differentiation, homeostasis, and pathogenesis.
Integrating multiple sources of information assumes a key role during these endeavors. Findings that uncover the involvement of epigenetic modifications in many biological processes, the complex regulatory pathways, and the crosstalk between them, are paving the way for one of the most exciting times in biology and medicine.
EpiTwin Project
One of the most ambitious projects to unveil epigenetic signatures is the EpiTwin project, initiated in 2010 as a collaborative endeavor between TwinsUK, at King’s College London, and the Beijing Genomics Institute.
TwinsUK, a nationwide registry in the U.K., was established in 1992 and currently has approximately 12,000 volunteer twins, most of them middle-aged and older, with an approximately equal number of monozygotic and dizygotic twins. “This is the largest epigenetic project of its kind,” says Tim Spector, M.D., professor of genetic epidemiology at King’s College London and director of the TwinsUK Registry.
Recently, in a search among TwinsUK participants for novel breast cancer-specific epigenetic biomarkers that could be detected in the blood, Dr. Spector and colleagues examined 30 monozygotic twin pairs discordant for breast cancer, and by high-resolution genome-wide DNA methylation profiling, unveiled over 400 differentially methylated genes.
“Sometimes this was apparent five years before diagnosis, showing that molecular changes occur years before clinical or biochemical changes can be examined,” says Dr. Spector.
These results pointed toward the hypermethylation of DOK7, which encodes the substrate and an activator of receptor tyrosine kinases, as either a potential candidate for the early blood-based diagnosis of this condition or as part of the early cancer process. The same gene was consistently hypermethylated in primary breast cancer and breast cancer cell lines.
“We believe that this setting allows us to make epigenetic discoveries in blood that are important in other tissues,” says Dr. Spector.

A recent epigenetics study explored breast cancer occurrence in twins discordant for the disease. Ultimately, the investigators identified over 400 differentially methylated genes. [Andrey Arkusha/Fotolia]

