
September 1, 2009 (Vol. 29, No. 15)
Steve Gardner, Ph.D.
Bioisostere Tools Aim to Make Optimized and Innovative Chemistry More Accessible
Modern biotech drug discovery often starts with the observation that the activity of a specific gene or protein is critical to the progression of a particular disease. Once confirmed in knock-down studies, hits can be identified by testing a small library of perhaps 500 compounds in an assay for activity against that target.
While these hits may have some activity, they are unlikely to have the mix of activity, specificity, and ADME/toxicity properties required to make a drug, and so the hits progress to the medicinal chemistry teams for hit to lead, lead optimization, and candidate selection. In smaller companies, these medicinal chemistry teams are often outsourced.
After receiving the preliminary hits, a medicinal chemist will take the 2-D structures of the hits in those screens and their activity data and create detailed structure activity relationships that attempt to explain the activity of the hits in terms of changes to their 2-D structure and other properties.
They will then attempt to predict, given knowledge gained from years of experience, which small evolutionary changes to that 2-D structure will enhance the affinity and specificity of the lead compound while overcoming any ADME and toxicity issues that might be observed. They will undertake iterative cycles of design, synthesis, and testing in an attempt to improve the molecular properties of the lead until it is optimized. This process can be time-consuming and expensive. During this process, the medicinal chemist’s reliance on insights gleaned from the 2-D structure may appear counterintuitive and confusing to many biologists—and they may be right to have reservations.
It has been known since the early days of drug discovery that molecules with different structural types (chemotypes) can have the same biological activity and properties. Such molecules are called bioisosteres. Moreover, x-ray crystallography has shown that these structurally diverse bioisosteres will bind to their common protein target in the same way. This similarity can be entirely independent of 2-D molecular structure, and the structural differences between bioisosteres can be so profound that it is impossible to predict by looking at them that their activities are related (Figure 1).
Because a protein target does not “see” the atoms and bonds of a drug, but instead interacts with the electron cloud around the molecule and the physicochemical properties at the surface of the compound, the 2-D structure is often a poor indicator of the biological activity and properties of a molecule.
Many bioisosteres have diverse chemotypes, well beyond the small degrees of 2-D structural difference that a medicinal chemist could possibly feel comfortable in suggesting as a change to a lead. This can create costly dead-ends or chemotype traps where the inability to predict a bioisosteric alternative to a given chemotype prevents the right mix of biological activity and properties from being achievable in a lead series.
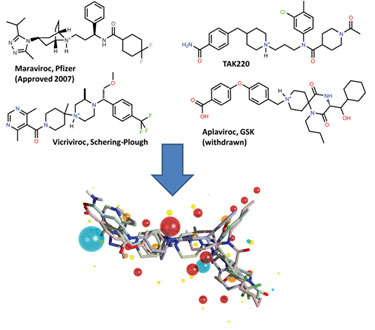
Figure 1. 3-D alignment of diverse CCR5 compounds sharing the same biological activity
Field-Based Approaches to Finding Bioisosteres
Modern computing has facilitated the development of a more accurate and biologically intuitive view of active molecules than the 2-D structures used by medicinal chemists. Field-based methods work by representing the chemical structure, not as a 2-D skeleton, but as a series of surface properties (molecular electrostatic potentials) that correspond to what the protein target actually interacts with when binding to a compound in its bioactive conformation.
Field-based tools mimic empirical observations of protein-drug interactions closely, and allow direct comparisons of the biologically important activities and properties of molecules completely independent of their 2-D structure.
Biologists can now use field-based tools to identify bioisosteres. Systems such as Cresset BioMolecular Discovery’s FieldStere are powerful enough to scan a wide range of chemical space and provide biologically intuitive results. Such tools can quickly generate a range of potential lead molecules directly from a set of initial hits with or without an x-ray structure of the target, where the lead molecule is free from known IP, ADME, or toxicity issues.
Using FieldStere is simple—enter an active hit structure, remove the portion of the molecule to be changed, specify any constraints on the type of chemistry required at the attachment points, and chose the fragment database to be searched. The results are automatically scored for similarity to the original compound and ranked in bioisosteric similarity order.
In scoring bioisosteres it is essential to remember that molecular fields are a property of the whole molecule and not of the isolated replacement fragment. Replacement fragments cannot be accurately assessed in isolation from the whole molecule as their properties can have a significant effect on the rest of the molecule. Scoring the whole molecule has an additional benefit in that the user is presented with a list of potentially active molecules with their full molecular properties such as rule-of-five violations and calculated LogP, which allows them to directly select molecules for synthesis more easily.
The originality and synthesizability of compounds produced using field technology is demonstrated in Figures 2 and 3. Using rofecoxib as a starting point, bioisosteres were sought that did not contain its lactone moiety. The chemistry space that was searched was limited by specifying that the replacement must contain an aromatic carbon attached to the remaining phenyl group. The results contain a range of bioisosteres from the reassuringly obvious such as Valdecoxib and Parecoxib to more interesting actives such as an analog of Etoricoxib. However, the results also contain nonobvious (and therefore potentially more innovative) bioisosteres such as the 12nM thiazolotriazole compound.
Accurate bioisostere identification tools such as FieldStere can shorten development time and increase the novelty of the lead series. It allows biologists working in companies without a large dedicated medicinal chemistry team to quickly gain insight into the diversity of the available active chemical space and provide much better chemistry starting points to their outsourcing partners. This in turn can reduce the number of wasted syntheses and increase the range of chemical and IP space explored.
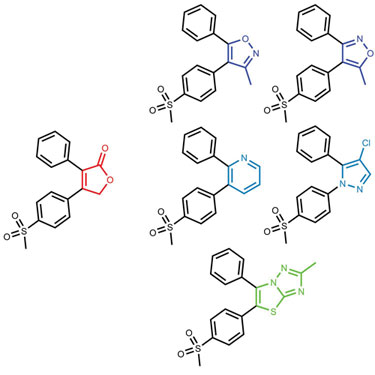
Figure 2. The results of a FieldStere experiment to find bioisosteres for the lactone portion of rofecoxib (red)
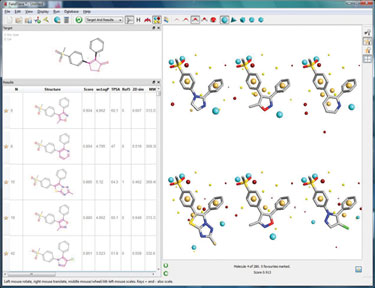
Figure 3. FieldStere screenshot showing the COX-2 results
Steve Gardner (steve@cresset-bmd. com) is COO at Cresset BioMolecular Discovery. Web: www.cresset-bmd.com.





