
March 1, 2013 (Vol. 33, No. 5)
With the tidal wave of next-generation sequencing sweeping through the landscape of biomedical sciences and technology, researchers are inundated with a flood of data.
Yet the data will only be as useful as the quality of the libraries from which it is generated, and can be no more informative than the use to which it is put.
Researchers at the “International Plant & Animal Genome” (PAG) conference presented their insights and opinions on such issues as how to prepare a DNA library from limited, degraded, or even an overabundance of material, as well as how best to piece together the information once it’s obtained.
When there is a lot of high-quality DNA to start with, it’s relatively easy to make a good sequencing library. Yet in too many cases only a limited quantity is available. New England Biolabs (NEB) has optimized its new NEBNext®Ultra DNA Library Prep Kit to address this issue—“you can use it with as little as 5 ng of human genomic DNA to make a library, and the quality of the library is very good,” said Pingfang Liu, Ph.D., application and product development scientist.
Another advantage for this kit is that it can be used with both intact DNA and fragmented DNA, like FFPE-preserved biopsies or circulating plasma DNA, she said, adding that other NGS library preparation kits on the market claiming facility with low DNA input require that DNA to be intact. “We have a lot of customers with precious samples, and they have only one shot to make a library.”
Being a company that specializes in enzymes, NEB focused on optimizing the enzymes and conditions found in the library prep kit. For example, it developed a polymerase for the PCR amplification with very good efficiency with all types of amplicons—including GC-rich and AT-rich—which boasts about 100-fold greater fidelity than Taq. The cleanup step between different enzyme reactions was also minimized, “because with a small amount of DNA, if you need to transfer from one tube to another tube you tend to lose a lot of your substrate,” Dr. Liu said.
The ligase master mix has been specially optimized to ligate the types of ends that are present in Illumina library prep methods, and at this point the kit is being supplied with adapters for Illumina library prep. It is compatible with the workflows of other sequencing platforms such as the Applied Biosystems’ 5500 Series SOLiD and Roche 454 platforms that use single-base TA overhangs as well, “although the sequences of the actual adapters are different between the different platforms,” she said.
Dr. Liu discussed results from a collaboration with Cynthia Hendrickson, Ph.D., of the Genomic Service Lab at HudsonAlpha Institute for Biotechnology, on exome capture. Using the NimbleGen SeqCapEZ Human Exome v3, Dr. Hendrickson was able to obtain virtually the same results starting with only 100 ng of genomic DNA using the NEBNextUltra DNA Library Prep Kit as with 2.5 µg genomic DNA using a traditional protocol.
This is all the more impressive when keeping in mind that “you will lose 99% of your library because the human exome is only 1% of the whole genome,” Dr. Liu remarked.
Divide, Skim, and Conquer
Ending up with only 1% of the genome has the obvious advantage of needing to sequence and analyze that much less DNA.
David Edwards, Ph.D., professor at the University of Queensland, used different means to address the challenges of the large and complex wheat genome, which is six times larger than the human genome. “It’s 80–90% repetitive. It contains three genomes—it’s hexaploid (as opposed to humans that are diploid). You can imagine the challenge of trying to sequence something like that!” he exclaimed.
One of Dr. Edwards’ collaborators isolates individual arms of chromosomes in microgram quantities, dissecting this complex genome into manageable pieces, reducing the complexity of assembling sequence data. “We’ve sequenced both arms of chromosome 7 from each of the three genomes now, and each one is the size of a rice genome,” he said.
Another way they reduce complexity is by using a skim-based genotyping-by-sequencing method—that is, sequencing at very low density, and calling the SNP where it matches a known polymorphism on the reference genome. “The advantage is that it’s essentially dial-able, you only need a very small amount of sequence data if you’re doing trait association,” he explained. “You only need very low coverage and it’s very cheap.”
Increasing the amount of data generated, on the other hand, yields a very high density of SNP genotypes, more than 3 million SNPs on chromosome 7 alone, which allows the group to examine the reference genome itself and compare haplotype blocks. If the haplotype block breaks down, part of the genome may have been mis-assembled or rearranged.
“So we’re using this high-density skim sequencing—that’s almost a contradiction in terms—to go through and validate and to fix genome assemblies,” Dr. Edwards said. “We’ve also used it for trait mapping, which is very straightforward and provides a physical rather than a genetic location, useful to identify candidate genes.”
They looked at the relationship among the three genomes (termed A, B, and D) to determine the impact that early farmers had on bread wheat. Most genes are conserved among the three genomes, and the differential gene loss that was found supported current theories of wheat’s evolution and domestication. The A and B genomes came together about 50,000 years ago—“the gene networks that are lost relate to it being grown in the wild,” Dr. Edwards explains.
The tetraploid wheat was then domesticated and dispersed by migration up through the Middle East into what is now southern Turkey where it came into contact with D genome wheat about 10,000 years ago. He said, “this was presumably growing in the same field as the domesticated tetraploid, and that formed a hybrid hexaploid wheat that became the bread wheat we eat today. The types of genes and the gene networks that are lost are really quite different, and this tells us that the new bread wheat was under very different selective pressure.”

Skim sequencing identifies high-resolution recombination events and misassembled reference genomes. Haplotype blocks are represented by vertical black bars, cultivar 1 is green, and cultivar 2 is purple. Each horizontal line represents a sibling of the F2-generation. [University of Queensland]
Pick and Choose
Sometimes sequencing the whole genome just gives more information than is really needed. If you want to focus on a particular set of genes or features in the genome, for example, it may be preferable to enrich for these ahead of time, saving the time, effort, and cost of a whole-genome analysis.
MYcroarray offers custom bait libraries designed to selectively capture DNA prior to sequencing. MYbaits target-enrichment kits can contain from hundreds to millions of 80 to 120 mer-long biotinylated RNA bait sequences. These are incubated together in liquid phase with the genomic sample for 36 hours, after which bound sample is pulled out of solution on streptavidin-coated magnetic beads. The baits are then degraded, leaving behind the positively selected DNA that can be used directly for, or amplified prior to, sequencing.
Bait sequences can be designed from genomic libraries or transcriptomes of the same or related species. “Say you’re working on mammals. You can eventually design your baits from the human genome, and then go to fish sequence from more distant genomes—it usually works well down to 90% sequence similarity or so,” said Jean-Marie Rouillard, Ph.D., CSO and co-founder of MYcroarray.
MYbaits is useful for gaining insights into gene structure, even when only transcriptome data is available. From transcriptomic baits the introns flanking the expressed sequences can be pulled down as well. It becomes possible to map exon-intron junctions, as well as to identify noncoding transcriptional and regulatory elements. This, in turn, will allow many fundamental phylogenetic and population questions to be addressed, Dr. Rouillard notes.
MYcroarray presented data in which a set of 14,468 bait sequences designed from western terrestrial garter snake transcriptome sequences were used to enrich genomic DNA prior to PCR amplification and 454 sequencing. 2,556 reference contigs were obtained: in these, 615,338 bases mapped to reference transcripts while 2,161,616 bases mapped to adjacent sequences, meaning that for each base of transcriptome sequence used for baiting an average 3½ bases of new sequence was discovered.
Another group presenting at PAG, led by Aaron Liston, Ph.D., professor at Oregon State University, used MYbaits to enrich for more than 6,000 previously identified polymorphic sites in strawberry, prior to genotyping of 48 F1 offspring. The resulting high-density linkage map allowed them to gain insights into sexual transitions of these and related diploid plant species.
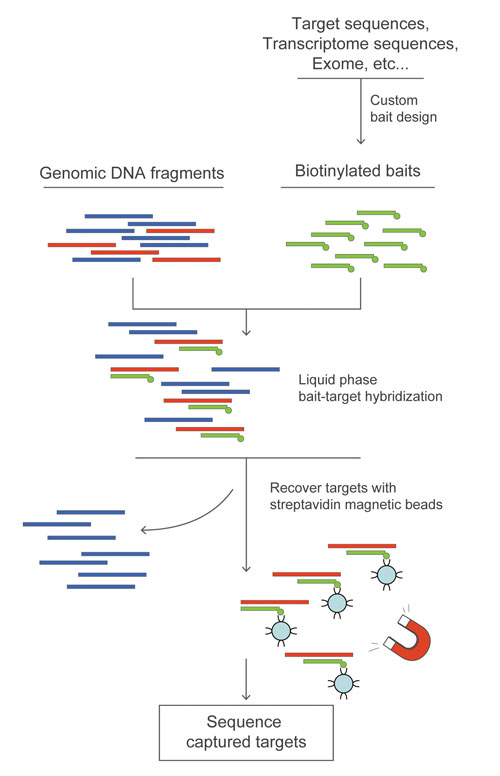
MYcroarray has created MYbaits custom biotinylated bait libraries for targeted sequencing.
Let’s Get Physical
Rather than looking at only selected portions of the genome to get a handle on assembling sequences, other approaches add complementary information to help piece together the data acquired from massively parallel next-gen sequencing reads of short DNA fragments.
The optical mapping of chromosomes has been around since at least 1993, when David Schwartz, Ph.D., and his colleagues described imaging of fluorescently labeled DNA molecules that had been digested with restriction enzymes after being fixed in agarose. The relative fluorescence intensity gave a measure of the length of each restriction fragment, and this information was used to construct ordered physical maps of the chromosomes from which they came.
OpGen took Dr. Schwartz’ technique into the realm of microfluidics, allowing whole-genome mapping (WGM) to be performed on its automated Argus platform. The Argus generates long single-molecule maps from 250 kilobases up to 2.5 megabases—giving “a more global picture,” and hence a more accurate analysis of structural variation such as indels, inversions, and translocations, said field application scientist Erin Newburn, Ph.D.
Smaller genomes can be mapped completely de novo by assembling the high-density restriction pattern repeats. But for larger genomes such as found in plants and animals, the company has introduced Genome-Builder™, a bioinformatics module “basically utilizing our very long restriction mapping leads of the WGM system and combining it with the sequencing scaffolds that our customers have assembled through their next-gen sequencing platforms,” she explained.
The genome sequence is converted to an in silico restriction map while at the same time long DNA molecules are isolated and cut by the Argus system. “Then we target the ends of these in silico maps and align up our single molecule restriction maps,” Dr. Newburn explained.
A subset of these will start to extend off the in silico map. After several iterations of alignments, extensions, and gap-bridging, “we can take these extended scaffolds (hybrid molecules, if you will) and then see if we can use traditional genetic information to connect the scaffolds and build what we’re calling a superscaffold.”
Dr. Newburn mentioned that a December article in Nature Biotechnology discussed the sequencing and mapping by a Chinese group of the domestic goat genome. By combining NGS with WGS they were able to significantly reduce the number of scaffolds, and they saw the N50 (a statistical measure of average length of the set of sequences) go from 3 megabases to 16.3 megabases—an improvement of more than fivefold, she related. “Many of the superscaffolds were actually on the order of the length of a full chromosome.”

